Adrenal gland diseases
Glucocorticoids
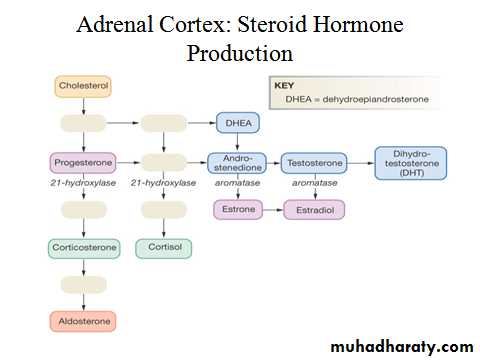
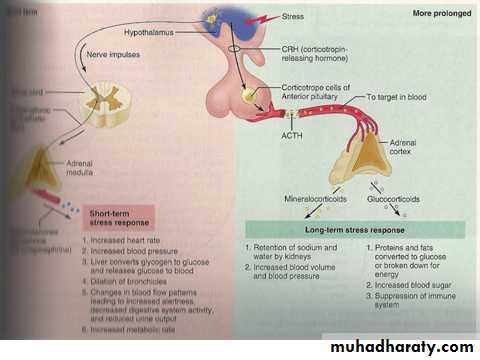
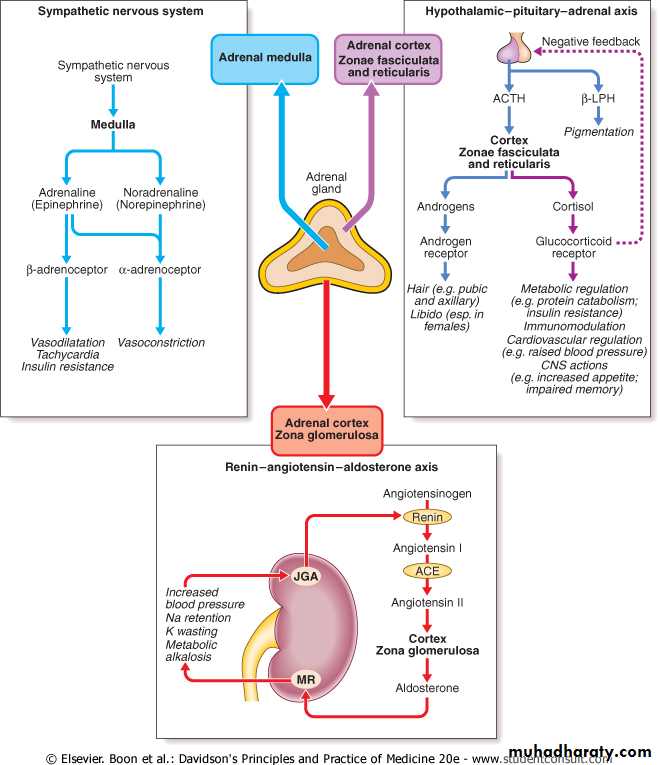
Cortisol is the major glucocorticoid in humans. Levels are highest in the morning on waking and lowest in the middle of the night. Cortisol rises dramatically during stress, including any illness. This elevation protects key metabolic functions (e.g. maintaining cerebral glucose supply during starvation) and puts an important 'brake' on potentially damaging inflammatory responses to infection and injury. The clinical importance of cortisol deficiency is, therefore, most obvious at times of stress.
Mineralocorticoids
Aldosterone is the body's most important sodium-retaining hormone, acting via mineralocorticoid receptors. Sodium is retained at the expense of increased excretion of potassium. Increased potassium in the lumen of the distal nephron also results in increased exchange with protons and metabolic alkalosis. The principal stimulus to aldosterone secretion is angiotensin II, a peptide produced by activation of the renin-angiotensin systemAdrenal androgens
Adrenal androgens are secreted in response to ACTH and are the most abundant steroids in the blood stream. They are probably important in the initiation of puberty (the adrenarche). The adrenals are also the major source of androgens in adult females and may be important in female libido.Catecholamines
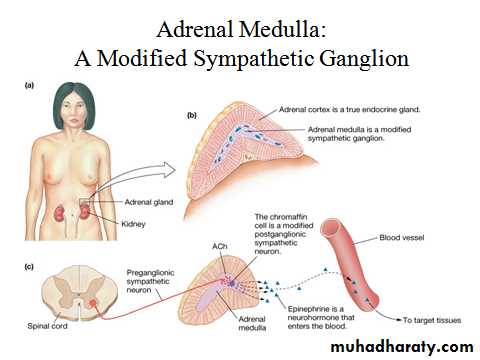
In humans, only a small proportion of circulating noradrenaline (norepinephrine) is derived from the adrenal medulla; much more is released from other nerve endings. The methyltransferase enzyme responsible for the conversion of noradrenaline to adrenaline (epinephrine) is induced by glucocorticoids. Blood flow in the adrenal is centripetal so that the medulla is bathed in high concentrations of cortisol and is the major source of circulating adrenaline.PHAEOCHROMOCYTOMA
This is a rare tumour of chromaffin tissue that secretes catecholamines and is responsible for less than 0.1% of cases of hypertension. There is a useful 'rule of tens' in this condition: ∼10% are malignant, ∼10% are extra-adrenal (i.e. elsewhere in the sympathetic chain) and ∼10% are familial.features
HTParoxysms…pallor , palpatation, sweating, anxiety
Abdominal pain and vomitting
Costipation, weight loss
Glucose intolerance
investigations
Measuring
Plasma….dopamin, adrenalin, noradrenalin
Urine…metabolite VMA, normetanephrine
Localisation
CT or MRI
Venous sampling with noradrenalin measurment
managment
Medical therapy to prepare patient for surgery for 6-8WAlpha blocker phenoxybenzamin better than prazocin (noncopetative inhibitor)
Combined alpha and beta labetalol
Na nitroprusside during surgery
Cushing syndrome
ClassificationACTH dependant
Pituitary adenoma secreting ACTH (i.e. Cushing's disease)
Ectopic ACTH syndrome (e.g. bronchial carcinoid, small-cell lung
carcinoma, pancreatic neuro-endocrine tumour)
latrogenic (ACTH therapy)
Non ACTH dependant
Iatrogenic (chronic glucocorticoid therapy, e.g. for asthma)
Adrenal adenoma
Adrenal carcinoma
Pseudo cushing Alcohol, depression, obesity
Clinical features
Hirsutism, hair thining, acne, moon face, straie, decrease skin thickness, bruising
PU, HT, DM, wasting , proximal myopathy, fractures, osteoporosis
Tendency for infection, poor wound healing
Menstural disturbance, psychosis, catract
investigations
Overnight low dose dexa sup or 24h urine free cortisol48h low dose dexa sup….confirm cushing
Plasma ACTH…not …adenoma
48h high dose dexa …supressed..pit…MRI
Not supress…ectopic ACTH(CXR)
Untreated cushinghave 50% 5 years mortality
Treatment is medical +surgicalMetyrapone, ketoconazole
Trans-sphenoidal adenectomy (microadenoma
Bilateral adrenalectomy
Theraputic uses of steroids
Side effects related to duration, dose, pre-existing conditionDouble dose in febrile illness
Add or increase dose in surgery or stress
Give IV if vomitting
Steroid cad or bracelet
Addisonian crisis
Shock, diarhea, vomitting, hypoglycemia, hypocalcemia, weakness.
Addisons disease
Inadequate secretion of stroid and or aldosterone, potentially fatal
Causes
Iatrogenic
Autoimmune
TB. HIV. Ca. surgical
features
Weakness, lethargy, wasting, anorexia, nausea , vomitting, diarrhea or constipation,Hypoglycemia, hypocalcemia, hyponatremia, hyperkalemia
Postural hypotension
Increase pigmentation
Decrease body hair, loss of lipido,
managment
Crisis…saline, GW, IV hydrocortisoneIdentify and treat underlying cause
Steroid replacement
mineralocorticoid
Hyperaldosteronism
Causes of mineralocorticoid excess*Secondary(high renin and aldosterone)
Diuretics, CHF, cirrhosis, nephrotic, RA stenosis
*primary(low renin, high aldosterone)
Adenoma (conns), bilateral hyperplasia
*low renin , low aldosterone
Ectopic ACTH, liquerce, liddles syn., CAH
features
Usually asymptomatic
Sodium retention or potassium loss, HT
Muscle weakness, polyuria, tetany, alkalosis
Na is high in primary low in secondary
Aldosteron;renin ratio
B-blockers inhibit thiazide stimulate renin
Ct or MRI for adenoma
Respond to spironolactone and amiloride.
CONGENITAL ADRENAL HYPERPLASIA
Aetiology and clinical featuresDefects in the cortisol biosynthetic pathway result in insufficiency of hormones 'distal' to the block, with impaired negative feedback and increased ACTH secretion. ACTH then stimulates the production of steroids 'proximal' to the enzyme block.
This produces adrenal hyperplasia and a combination of clinical features that depend on the severity and site of the defect in biosynthesis. All of these enzyme abnormalities are inherited as autosomal recessive traits.
The most common enzyme defect is 21-hydroxylase deficiency
This results in impaired synthesis of cortisol and aldosterone and accumulation of 17OH-progesterone, which is then diverted to form adrenal androgens
Features
Glucocorticoid or mineralocorticoid deficiencyAmbigous genetali in girls, precocious puberty
Amenorrhea, hirsutism
Management
The aim is to replace deficient corticosteroids, and also suppress ACTH and hence adrenal androgen production. In contrast with glucocorticoid replacement therapy in other forms of cortisol deficiency , it is usual to give 'reverse' treatment, i.e. a larger dose of a long-acting synthetic glucocorticoid just before going to bed to suppress the early morning ACTH peak, and a smaller dose in the morning.
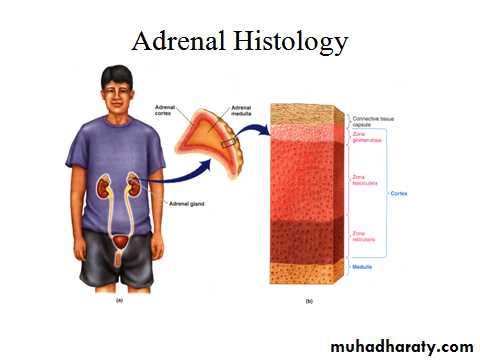
ADRENALS
Glucocorticoids
Cortisol is the major glucocorticoid in humans. Levels are highest in the morning on waking and lowest in the middle of the night. Cortisol rises dramatically during stress, including any illness. This elevation protects key metabolic functions (e.g. maintaining cerebral glucose supply during starvation) and puts an important 'brake' on potentially damaging inflammatory responses to infection and injury. The clinical importance of cortisol deficiency is, therefore, most obvious at times of stress.Mineralocorticoids
Aldosterone is the body's most important sodium-retaining hormone, acting via mineralocorticoid receptors. Sodium is retained at the expense of increased excretion of potassium. Increased potassium in the lumen of the distal nephron also results in increased exchange with protons and metabolic alkalosis. The principal stimulus to aldosterone secretion is angiotensin II, a peptide produced by activation of the renin-angiotensin system). Renin secretion from the juxtaglomerular apparatus in the kidney is stimulated by low perfusion pressure in the afferent arteriole, low sodium filtration leading to low sodium concentrations at the macula densa, or increased sympathetic nerve activity.Catecholamines
In humans, only a small proportion of circulating noradrenaline (norepinephrine) is derived from the adrenal medulla; much more is released from other nerve endings. The methyltransferase enzyme responsible for the conversion of noradrenaline to adrenaline (epinephrine) is induced by glucocorticoids. Blood flow in the adrenal is centripetal so that the medulla is bathed in high concentrations of cortisol and is the major source of circulating adrenaline. However, in the absence of functioning adrenal medullae, e.g. after bilateral adrenalectomy, there appear to be no clinical consequences attributable to deficiency of circulating catecholamines.Adrenal androgens
Adrenal androgens are secreted in response to ACTH and are the most abundant steroids in the blood stream. They are probably important in the initiation of puberty (the adrenarche). The adrenals are also the major source of androgens in adult females and may be important in female libido.CUSHING SYNDROME
PRESENTING PROBLEMS IN ADRENAL DISEASE
ACTH-dependentPituitary adenoma secreting ACTH (i.e. Cushing's disease)
Ectopic ACTH syndrome (e.g. bronchial carcinoid, small-cell lung
carcinoma, pancreatic neuro-endocrine tumour)
latrogenic (ACTH therapy)
Non-ACTH-dependent
Iatrogenic (chronic glucocorticoid therapy, e.g. for asthma)
Adrenal adenoma
Adrenal carcinoma
Pseudo-Cushing's syndrome, i.e. cortisol excess as part of another illness Alcohol excess (biochemical and clinical features),Major depressive illness(biochemical) and obesity(clinical)
Features
SymptomsTrouble sleeping-trouble falling asleep or frequent awakenings
Severe fatigue-new onset
Abrupt weight gain-without other cause such as decreased activity or depression
Menstrual abnormalities
Cognitive changes- “brain fog”
Decreased Libido
Depression, anxiety, mood-swings
Signs
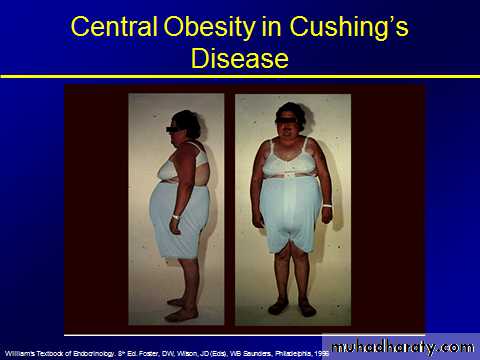
Central obesity
Muscle atrophy
Thin skin
Buffalo hump
Round, red face
Bruising
Extra hair growth
Acne
Loss of hair on head
Stretch marks
Clinical assessment
spontaneous Cushing's syndrome is rare, the positive predictive value of any one feature alone is low. Moreover, some common disorders can be confused with Cushing's syndrome because they are associated with alterations in cortisol secretion: for example, obesitythe best predictive value in favour of Cushing's syndrome in an obese patient are bruising, myopathy and hypertension.
In all patients with features of Cushing's syndrome it is vital to exclude iatrogenic causes even inhaled or topical glucocorticoid
Unlike pituitary tumours secreting ACTH, ectopic tumours have no residual negative feedback sensitivity to cortisol, and both ACTH and cortisol levels are usually higher than with other causes. Very high ACTH levels are associated with marked pigmentation. Very high cortisol levels overcome the barrier of 11β-HSD2 in the kidney and cause hypokalaemic alkalosis.
When the tumour secreting ACTH is malignant (e.g. small-cell lung carcinoma), then the onset is usually rapid and may be associated with cachexia.
In Cushing's disease, the pituitary tumour is usually a microadenoma (< 10 mm in diameter); hence other features of a pituitary macroadenoma (hypopituitarism, visual failure or disconnection hyperprolactinaemia,
In iatrogenic Cushing's syndrome, cortisol levels are low unless the patient is taking a corticosteroid (such as prednisolone) which cross-reacts in immunoassays with cortisol. Plasma cortisol levels are highly variable in healthy subjects so that patients with Cushing's syndrome often have daytime values within the normal range. For this reason,
there is no place for a random measurement of daytime plasma cortisol in the clinic in either supporting or refuting the diagnosis.
Cushing's syndrome is confirmed by the demonstration of increased secretion of cortisol (measured in urine) that fails to suppress with relatively low doses of dexamethasone (measured in plasma or urine)
Dexamethasone is used for suppression testing because, unlike prednisolone, it does not cross-react in radioimmunoassays for cortisol.
Once the presence of Cushing's syndrome is confirmed, measurement of plasma ACTH is the key to establishing the differential diagnosis. In the presence of excess cortisol secretion, an undetectable ACTH indicates an adrenal tumour, while any detectable ACTH is pathological.
Tests to discriminate pituitary from ectopic sources of ACTH rely on the fact that pituitary tumours, but not ectopic tumours, retain some features of normal regulation of ACTH secretion. Thus, in Cushing's disease ACTH secretion is suppressed by dexamethasone, albeit at a higher dose than in health, and ACTH is stimulated by corticotrophin-releasing hormone (CRH).
Management
Untreated Cushing's syndrome has a 50% 5-year mortality. Most patients are treated surgically with medical therapy given for a few weeks prior to operation.A number of drugs are used to inhibit corticosteroid biosynthesis, including metyrapone, aminoglutethimide and ketoconazole. The dose of these agents is best titrated against 24-hour urine free cortisol.
Trans-sphenoidal surgery with selective removal of the adenoma is the treatment of choice.
If bilateral adrenalectomy is used in patients with pituitary-dependent Cushing's syndrome, then there is a risk that the pituitary tumour will grow in the absence of the negative feedback suppression previously provided by elevated cortisol levels. This can result in Nelson's syndrome
Adrenal adenomas are removed via laparoscopy or a loin incision
ADRENAL INSUFFICIENCY
CAUSES OF ADRENOCORTICAL INSUFFICIENCYSecondary (↓ACTH)
Withdrawal of suppressive glucocorticoid therapy
Hypothalamic or pituitary disease
Primary (↑ACTH) Common causes
Autoimmune
Sporadic
Polyglandular syndromes before administering hydrocortisone, but investigations may need to be delayed until after recovery.
features
SymptomsHyperpigmentation of the skin and mucous membranes
Vitiligo
Dizziness with orthostasis
Myalgias and flaccid muscle paralysis
progressive weakness, fatigue, poor appetite, and weight loss.
gastrointestinal symptoms
Signs
dehydration, hypotension, and orthostasis.increased pigmentation of the skin and mucous membranes, with or without areas of vitiligo.
absence of axillary and pubic hair and decreased body hair.
Wasting, proximal myopathy
hyponatremia, hyperkalemia, and a mild non–anion-gap metabolic acidosiselevated blood urea nitrogen (BUN) and creatinine due to the hypovolemia,Hypoglycemia
Assessment of glucocorticoids
Random plasma cortisol is usually low in patients with adrenal insufficiency, but it may be within the normal range yet inappropriately low for a seriously ill patient.ACTH STIMULATION TEST
Diagnosis of primary or secondary adrenal insufficiency
Assessment of hypothalamic-pituitary-adrenal axis in patients taking suppressive glucocorticoid therapy
Relies on ACTH-dependent adrenal atrophy in secondary adrenal insufficiency, so may not detect acute ACTH deficiency (e.g. in pituitary apoplexy. Cortisol levels fail to increase in response to exogenous ACTH in patients with primary or secondary adrenal insufficiency.
If an ACTH assay is unavailable, then a long ACTH stimulation test can be used (1 mg depot ACTH i.m. daily for 3 days); in secondary adrenal insufficiency there is a progressive increase in plasma cortisol with repeated ACTH administration, whereas in Addison's disease cortisol remains less than 700 nmol/l (25.4 μg/dl) at 8 hours after the last injection.
All glucocorticoid therapy, even if inhaled or applied topically, can suppress the hypothalamic-pituitary-adrenal axis (HPA). In practice, this is only likely to result in a crisis due to adrenal insufficiency on withdrawal of treatment if glucocorticoids have been administered orally or systemically for longer than 3 weeks, if repeated courses have been prescribed within the previous year, or if the dose is higher than the equivalent of 40 mg prednisolone per day.
In these circumstances, the drug, when it is no longer required for the underlying condition, must be withdrawn slowly at a rate dictated by the duration of treatment. If glucocorticoid therapy has been prolonged, then it may take many months for the HPA to recover.
Recovery of the HPA is aided if there is no exogenous glucocorticoid present during the nocturnal surge in ACTH secretion, i.e. if the glucocorticoid is given in the morning. Giving ACTH to stimulate adrenal recovery is of no value as the pituitary remains suppressed.
patients who have received glucocorticoids for longer than a few weeks, it is often valuable to confirm that the HPA is recovering during glucocorticoid withdrawal. Once the dose of glucocorticoid is reduced to a minimum (e.g. 4 mg prednisolone or 0.5 mg dexamethasone per day), then measure plasma cortisol at 0900 hrs before the next dose. If this is detectable, then perform an ACTH stimulation test
In addisons replacment therapy
hyperaldosteronism
The prevalence of primary hyperaldosteronism is controversial. If only hypertensive patients with hypokalaemia are investigated, then fewer than 1% of patients with hypertension will be found to have primary hyperaldosteronism. Around half of these have an adrenal adenoma secreting aldosterone (Conn's syndrome). However, recent studies in which hypertensive patients have been screened using aldosterone/renin ratios (see below) suggest that the prevalence may be as high as 5%. Most of these 'extra' patients have bilateral adrenal hyperplasia rather than Conn's syndrome and many have normal plasma potassium. Although mineralocorticoid receptor antagonists might be the antihypertensive agent of choice in such patients, it remains to be determined whether investigation of all hypertensive patients for bilateral adrenal hyperplasia is worthwhile.Secondary hyperaldosteronism may be associated with hypertension in renovascular disease and in very rare renin-secreting renal tumours. Renal artery stenosis, Bartter syndrome, pregnancy, heart failure, cirrhosis.
Management; spironolactone, surgery
PHAEOCHROMOCYTOMA
CLINICAL FEATURES OF PHAEOCHROMOCYTOMAHypertension (usually paroxysmal; often postural drop of blood pressure)
Paroxysms of:
Pallor (occasionally flushing)
Palpitations
Sweating
Headache
Anxiety (fear of death-angor animi)
Abdominal pain, vomiting
Constipation
Weight loss
Glucose intolerance
investigations
Excessive secretion of catecholamines can be confirmed by measuring the hormones (adrenaline/epinephrine, noradrenaline/norepinephrine and dopamine) in plasma or their metabolites (e.g. vanillyl-mandelic acid, VMA; conjugated metanephrine and normetanephrine) in urine. However, catecholamine secretion is usually paroxysmal and sometimes the paroxysms are infrequent. Therefore, false-negative results may be obtained if samples are collected during a period when symptoms or hypertension are absent.Phaeochromocytomas are usually identified by abdominal CT or MRI . Difficulty can arise with the localisation of extra-adrenal tumours. Scintigraphy using meta-iodobenzyl guanidine (MIBG) can be useful. Selective venous sampling with measurement of plasma noradrenaline (norepinephrine) may be required.
Managment
Medical therapy is required to prepare the patient for surgery, preferably for a minimum of 6 weeks to allow restoration of normal plasma volume. The most useful drug in the face of very high circulating catecholamines is the α-blocker phenoxybenzamine (10-20 mg orally 6-8-hourly) because it is a non-competitive antagonist, unlike prazosin or doxazosin. If α-blockade produces a marked tachycardia, then a β-blocker (e.g. propranolol) or combined α- and β-antagonist (e.g. labetalol) can be added. On no account should the β-antagonist be given before the α-antagonist, as it may cause a paradoxical rise in blood pressure due to unopposed α-mediated vasoconstriction.During surgery sodium nitroprusside and the short-acting α-antagonist phentolamine are useful in controlling hypertensive episodes which may result from anaesthetic induction or tumour mobilisation.
CONGENITAL ADRENAL HYPERPLASIA
FeaturesDefects in the cortisol biosynthetic pathway result in insufficiency of hormones 'distal' to the block, with impaired negative feedback and increased ACTH secretion. ACTH then stimulates the production of steroids 'proximal' to the enzyme block. This produces adrenal hyperplasia and a combination of clinical features that depend on the severity and site of the defect in biosynthesis.
The most common enzyme defect is 21-hydroxylase deficiency. This results in impaired synthesis of cortisol and aldosterone and accumulation of 17OH-progesterone, which is then diverted to form adrenal androgens
High levels of plasma 17OH-progesterone are found in 21-hydroxylase deficiency. In late-onset cases this may only be demonstrated after ACTH administration. To avoid salt-wasting crises in infancy, 17OH-progesterone can be routinely measured in heel prick blood spot samples taken from all infants in the first week of life.
Managment
The aim is to replace deficient corticosteroids, and also suppress ACTH and hence adrenal androgen production. In contrast with glucocorticoid replacement therapy in other forms of cortisol deficiency , it is usual to give 'reverse' treatment, i.e. a larger dose of a long-acting synthetic glucocorticoid just before going to bed to suppress the early morning ACTH peak, and a smaller dose in the morning. A careful balance is required between adequate suppression of adrenal androgen excess and excessive glucocorticoid replacement resulting in features of Cushing's syndrome.In adults, clinical features (menstrual cycle, hirsutism, weight gain, blood pressure) and biochemical profiles (plasma renin activity and 17OH-progesterone levels) provide a guide.
late-onset 21-hydroxylase deficiency may not require corticosteroid replacement. If hirsutism is the main problem, anti-androgen therapy may be just as effective