Bronchiectasis
Dr .Ghazi F.HajiIntervention cardiology
Al- kindi medical college
Pathology
The bronchiectatic :cavities may be lined by granulation tissue, squamous epithelium or normal ciliated epithelium. There may also be inflammatory changes in the deeper layers of the bronchial wall and hypertrophy of the bronchial arteries.
resulting in progressive destruction of the normal lung architecture in advanced cases.
Causes of bronchiectasis
CongenitalCystic fibrosis
Ciliary dysfunction syndromes
Primary ciliary dyskinesia (immotile cilia syndrome)
Kartagener's syndrome (sinusitis and transposition of the viscera)
Primary hypogammaglobulinaemia
Acquired: children
Pneumonia (complicating whooping cough or measles)
Primary TB
Inhaled foreign body
Acquired: adults
Suppurative pneumonia
Pulmonary TB
Allergic bronchopulmonary aspergillosis complicating asthma
Bronchial tumours
Clinical features Symptoms of bronchiectasis
Cough Chronic productive cough ,usually worse in mornings and often brought on by changes of posture. Sputum often copious and persistently purulent in advanced disease. Halitosis is a common accompanying feature
Pneumonia and pleurisy : fever, malaise with pleurisy.
Haemoptysis Can be slight or massive and is often recurrent. '
Poor general health ,there may be associated weight loss, anorexia, lassitude, low-grade fever, and failure to thrive in children.
Digital clubbing is common
Physical signs
May be no abnormal physical signs.-coarse crackles may be heard over the affected areas.
-Diminished breath sounds due to Collapse
-Bronchial breathing if consolidation occur.
-Pleural rub if pleurisy occur.
Investigations
1-Bacteriological and mycological examination of sputum(sputum culture )Pseudomonas aeruginosa, fungi such as Aspergillus and various mycobacteria.
2-Radiological examination
Bronchiectasis, may be normal chest X-ray.
In advanced disease,cystic bronchiectatic spaces, pneumonic consolidation or collapse may be visible.
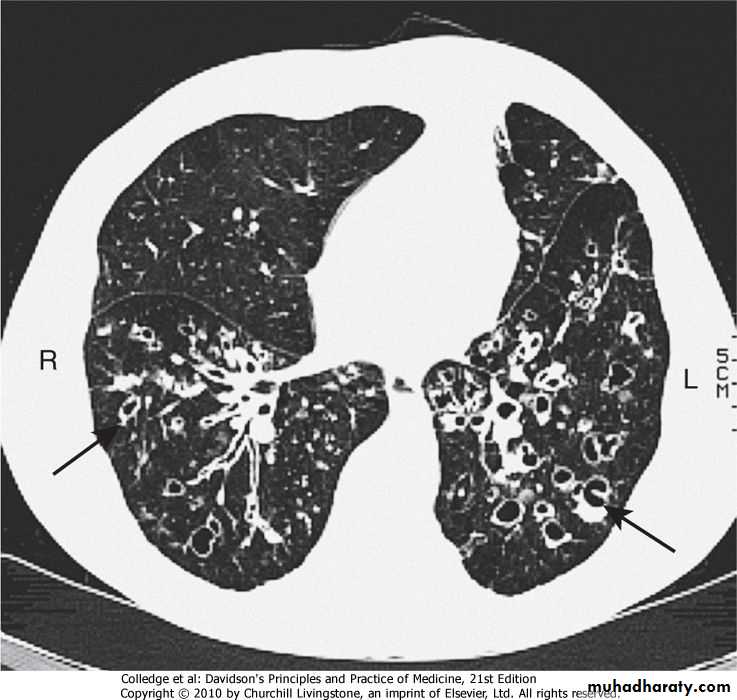
3-CT is much more sensitive, and shows thickened dilated airways
4-Assessment of ciliary functionA screening test can be performed in patients suspected of having a ciliary dysfunction syndrome by measuring the time taken for a small pellet of saccharin placed in the anterior chamber of the nose to reach the pharynx, when the patient can taste it. This time should not exceed 20 minutes but is greatly prolonged in patients with ciliary dysfunction.
Management
1-Physiotherapy-Patients should be instructed on how to perform regular daily physiotherapy to assist the drainage of excess bronchial secretions.
-The optimum duration and frequency of physiotherapy depend on the amount of sputum, but 5-10 minutes once or twice daily is a minimum for most patients.
In patients with airflow obstruction,2- inhaled bronchodilators and corticosteroids should be used to enhance airway patency.
3-Antibiotic therapy
staphylococci and Gram-negative bacilli(Pseudomonas species),
oral ciprofloxacin (250-750 mg 12-hourly)
or ceftazidime by intravenous injection
or infusion (1-2 g 8-hourly) may be required.
Surgical treatment
Excision of bronchiectatic areas is only indicated in a small proportion of cases.
In progressive forms of bronchiectasis, resection of destroyed areas of lung which are acting as a reservoir of infection should only be considered as a last resort.
Prognosis
The disease is progressive when associated with ciliary dysfunction and cystic fibrosis, and eventually causes respiratory failure.In other patients the prognosis can be relatively good if physiotherapy is performed regularly and antibiotics are used aggressively.
Prevention
As bronchiectasis commonly starts in childhood following measles, whooping cough or a primary tuberculous infection, it is essential that these conditions receive adequate prophylaxis and treatment. The early recognition and treatment of bronchial obstruction is also importantCystic fibrosis(CF)
@Cystic fibrosis (CF) is the most common fatal genetic disease in Caucasians, with autosomal recessive inheritance, an incidence of about 1 in 2500 live births .@CF is the result of mutations affecting a gene on the long arm of chromosome 7 which codes for a chloride channel known as cystic fibrosis transmembrane conductance regulator (CFTR), that influences salt and water movement across epithelial cell membranes.
.
$The genetic defect causes increased sodium and chloride content in sweat and increased resorption of sodium and water from respiratory epithelium .
Relative dehydration of the airway epithelium is thought to predispose to chronic bacterial infection and ciliary dysfunction, leading to bronchiectasis.
$The gene defect also causes disorders in the gut epithelium, pancreas, liver and reproductive tract
#In the 1960s, few patients with CF survived childhood, yet with aggressive treatment of airway infection and nutritional support, life expectancy has improved dramatically, such that there are now more adults than children with CF in many developed countries.
#Prenatal screening by amniocentesis may be offered to those known to be at high risk.
Clinical features@Recurrent exacerbations of bronchiectasis(signs and symptoms). Common Infections with
( Staph. Aureus,Pseudomonas species Aspergillus and 'atypical mycobacteria) in CF
@Most men with CF are infertile due to failure of development of the vas deferens
@Genotype is a poor predictor of disease severity in individuals; even siblings with matching genotypes may have quite different phenotypes.
Complications of cystic fibrosis
RespiratoryInfective exacerbations of bronchiectasis
Spontaneous pneumothorax
Haemoptysis /Nasal polyps
Respiratory failure /Cor pulmonale
Lobar collapse due to secretions
Gastrointestinal
Malabsorption and steatorrhoea /Distal intestinal obstruction syndrome
Biliary cirrhosis and portal hypertension /Gallstones
Others Diabetes (25% of adults) /Delayed puberty /Male infertility
Stress incontinence due to repeated forced cough /Psychosocial problemsOsteoporosis /Arthropathy
Cutaneous vasculitis
Management
1-Regular chest physiotherapyTO reduce chest exacerbations and/or improve lung function in CF
. 2- Antibiotic: Infections with( Staph. Aureus,Pseudomonas species Aspergillus and 'atypical mycobacteria)1-Nebulised tobramycin 300 mg 12-hourly, given in alternate months
2-Regular oral azithromycin 500 mg three times/weeK3-Nebulizer recombinant human DNase 2.5 mg daily .
4-Some patients have coexistent asthma, which is treated with inhaled bronchodilators and corticosteroids;
5-For advanced CF lung disease, home oxygen and NIV may be necessary to treat respiratory failure.
6-lung transplantation can produce dramatic improvements but is limited by donor organ availability.
7-Somatic gene therapy:
@The discovery of the CF gene is located in the respiratory epithelium (which is accessible by inhaled therapy).
@Manufactured normal CF gene can be 'packaged' within a viral or liposome vector and delivered to the respiratory epithelium to correct the genetic defect.
@Initial trials in the nasal and bronchial epithelium have shown some effect, and further trials of nebulised bronchial delivery are planned..