
123
The hemoglobin disorders
Genetic disorders of human hemoglobin are the most common group of single- gene
diseases mutation, estimated 7% of the world's population carries one or more
mutations of the genes involved in hemoglobin synthesis.
The hemoglobin molecule
The hemoglobin molecule is a tetramer composed of four
polypeptide chains, two α and two β.
The ß chains are encoded by a gene on chromosome 11, and
the α chains are encoded by two genes on chromosome 16.
A normal individual has two normal β genes and four
normal α genes. Each of these globin chains is associated
with a heme group, which contains an iron atom and
binds with O2.
The hemoglobin disorders can be classified into two broad categories:
Structural abnormalities: in which the Hb
molecule is altered (ex. Sickle cell disease),
and
thalassemias: a group of conditions in which
either the α- or the β- globin chain is structurally
normal but reduced in quantity.
Hereditary persistence of fetal hemoglobin (HPFH),
occurs when fetal hemoglobin, encoded by the α-
globin genes and by two β- globin- like genes called
A
γ and
G
γ continue to produces after birth (normally
γ- chain production ceases and β- chain production
begins at time of birth). HPFH does not cause
disease.
124
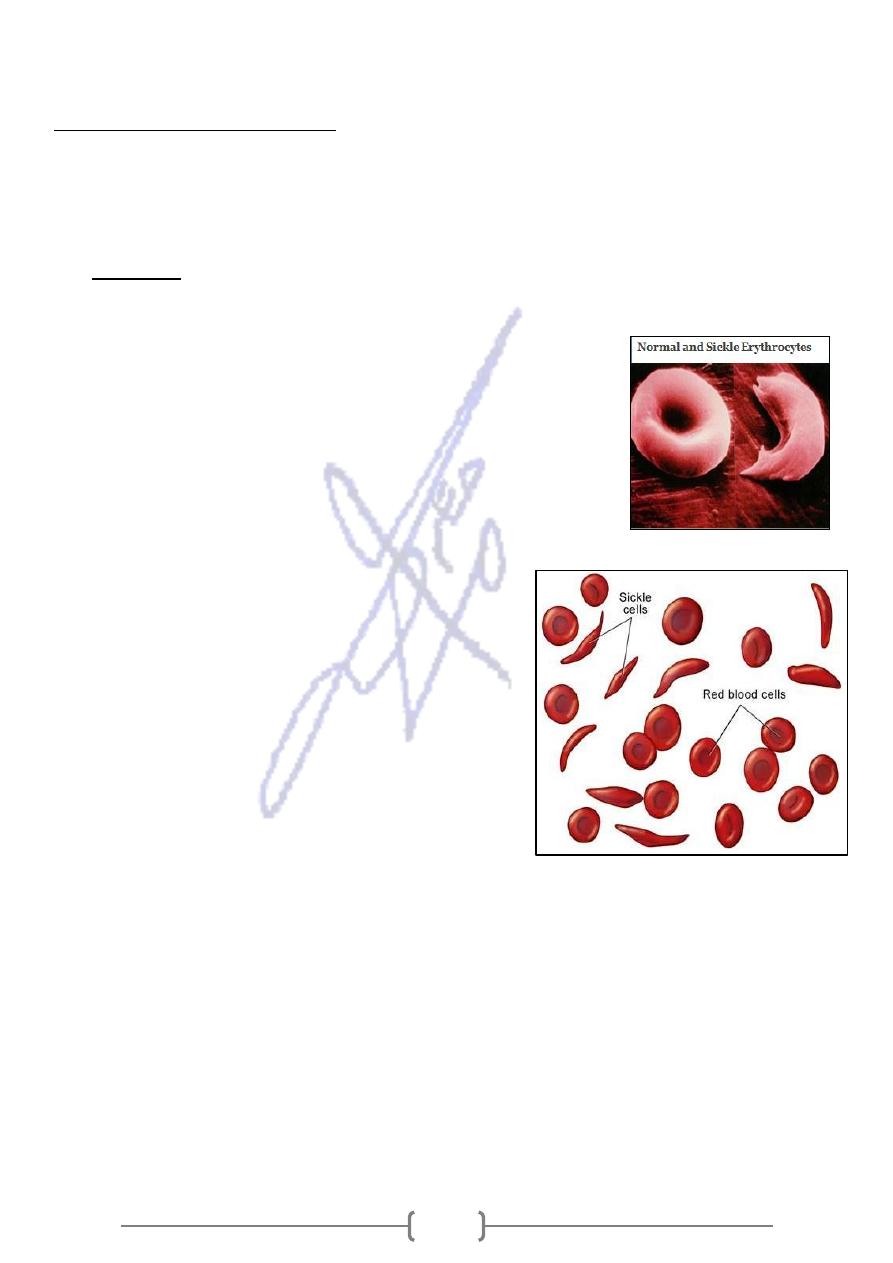
1. Sickle Cell Disease (SCD):
It is a structural hemoglobin disorder, affects mostly African 1 in 50 births, African –
American birth 1 in 400 births, and occasionally in Mediterranean and middle eastern
populations.
a single missense mutation that affects the substitution of valine for
caused by
It
glutamic acid at position 6 of the β- globin polypeptide chain.
In homozygous form (HbSS), the most common form, this
mutation alter the characteristics of the hemoglobin
molecule. Sickle hemoglobin, has a tendency to polymerize
into rod-like structures that alter the shape of the usually
flexible red blood cells, The cells take on a shape that
resembles the curved blade of the sickle under the
condition of low oxygen tension.
Affected individuals present with a wide range of
clinical problems that result from vascular
obstruction and hypoxemia (because the sickle
erythrocytes are less flexible and tend to stick to
the vascular endothelium).
Premature destruction of the sickled erythrocytes
resulting in chronic anemia.
Although the disease can be diagnosed at birth,
clinical abnormalities usually do not occur before
age 6 months
The spleen becomes enlarged and destroyed by
infarctions.
Recurrent bacterial infection, and frequently cause of death.
Carriers of the sickle cell gene are said to have sickle cell trait (Heterozygous)
Unlike sickle cell disease, sickle cell trait does not cause health problems.
When both parents are carriers of sickle cell trait, there is a 25% chance in each
pregnancy for the baby to inherit two sickle cell genes and have sickle cell anemia
(HbS), or SS disease.
Correspondingly, there is a 50% chance the baby will have sickle cell trait (carrier)
and a 25% chance that the baby will have the usual type of hemoglobin (HbA).

125
2. Thalassemia
The term “thalassemia” is derived from the Greek word thalassa (sea).
Thalassemia can be divided into two major groups:
globin chain
thalassemia depending on the
-
β
thalassemia and
-
α
quantity.
that is reduced in
When one type of chain is decreased in number, the other chain
to participate in normal tetramer formation and tend to
unable
type
form molecules consisting of four chains of the excess type only
(homotetramers, in contrast to the heterotetramers normally
formed by α and β chains).
thalassemia
-
α
Most case of α- thalassemia are caused by deletion of the α- globin genes.
The loss of one gene "silent" carrier. This means you has no clinical effect.
The loss of two genes, you have α- thalassemia trait (also called α- thalassemia
minor). You may have mild anemia.
The loss or abnormality of three of the α- genes, will produce hemoglobin H disease
(HbH). This form of thalassemia causes moderate to severe anemia.
Very rarely, a baby will be missing all four genes. This condition is called α-
thalassemia major or hydrops fetalis. Babies who have hydrops fetalis usually die
before or shortly after birth.
thalassemia
-
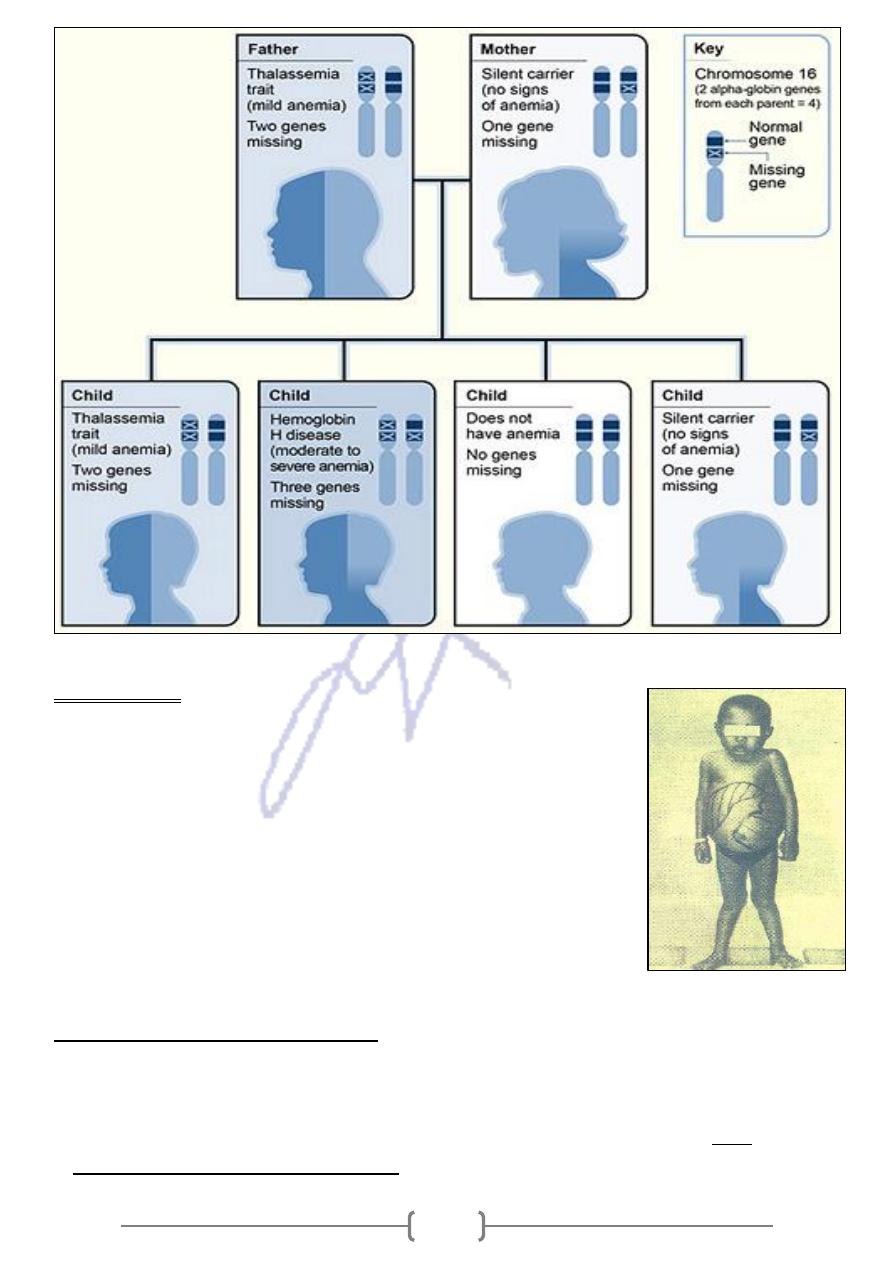
Inheritance Pattern for α
The picture shows how α- thalassemia is inherited:
The α- globin genes are located on chromosome 16.
father
e, the
two from each parent. In this exampl
-
globin genes
-
α
A child inherits four
.
globin gene
-
α
mother is missing one
genes and the
globin
-
α
is missing two
Each child has a 25% chance of inheriting two missing genes and two normal genes
(thalassemia trait),
three missing genes and one normal gene (hemoglobin H disease),
four normal genes (no anemia), or
one missing gene and three normal genes (silent carrier).
126
thalassemia
-
β
Individuals with a β- globin mutation in one copy of chromosome
11 (heterozygous) are said to have β- thalassemia minor. It
causes mild anemia and do not required clinical management.
Those with both copies of the chromosome carry a β-globin
mutation (Homozygous) develop either
1) β- thalassemia major (also called Cooley's anemia) which
causes severe anemia.
2) or the less serious condition,
β- thalassemia intermedia causes moderate anemia.
thalassemia
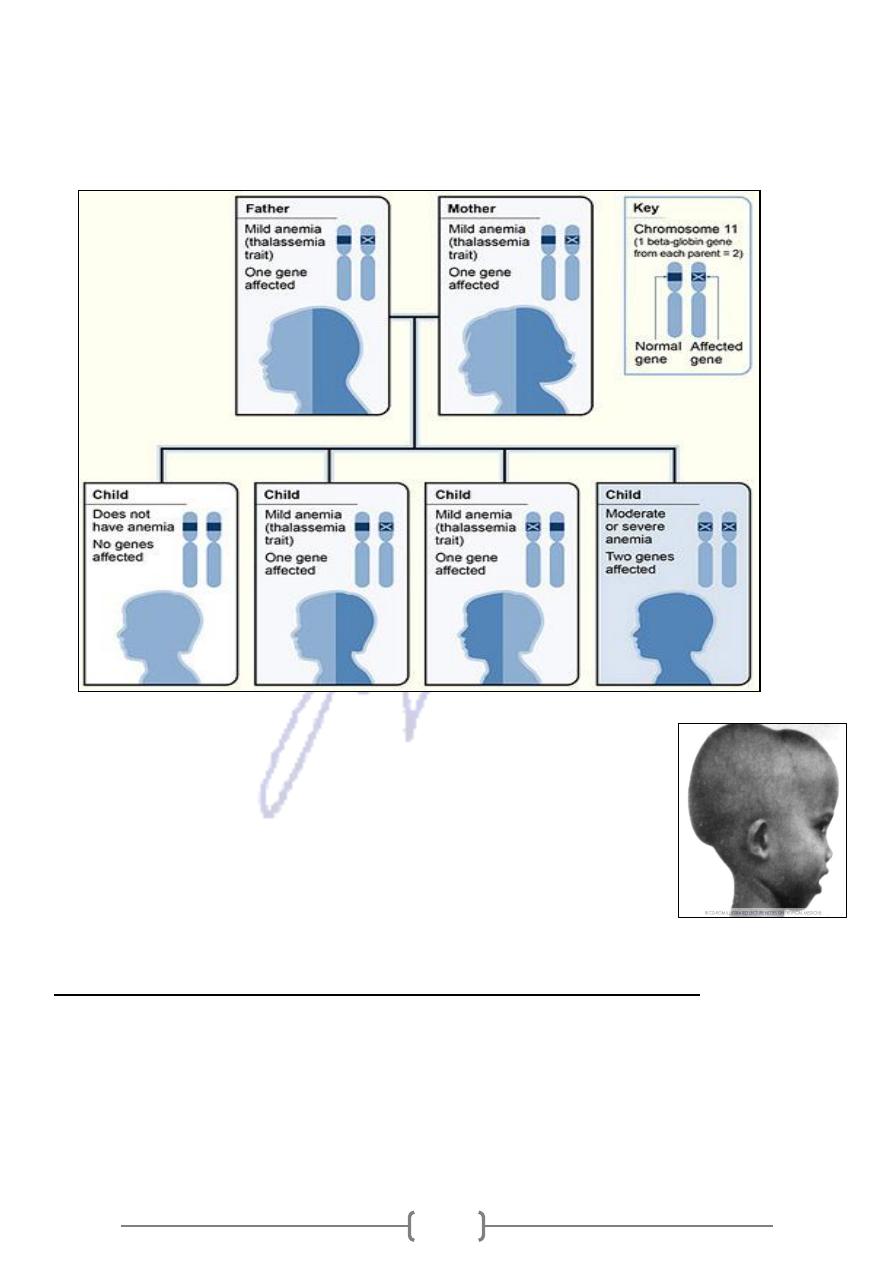
-
Inheritance Pattern for β
The picture shows how β- thalassemia is inherited.
The beta globin gene is located on chromosome 11.
each
one from each parent. In this example,
-
globin genes
-
A child inherits two β
globin gene.
-
parent has one altered β
127
Each child has a 25% chance of inheriting two normal genes (no anemia),
a 50% chance of inheriting one altered gene and one normal gene (β- thalassemia
trait or minor), or
a 25% chance of inheriting two altered genes (beta thalassemia major).
Β-globin is not produced until after birth, so the effect of β-
thalassemia major are not seen clinically until the age 2-6 months
The anemia causes bone marrow expansion, which in turn produces
skeletal changes, including a protuberant upper jaw and cheek-
bones and thinning of the long bones.
Splenomegaly and infection are common.
Without treatment patient may often die during the first decade of
life.
includes:
base mutations
-
single
thalassemia are caused by
-
β
of
Most case
1. Nonesense mutation: result in premature termination of translation of the β- globin
chain (completely absent of β- globin)
2. Frameshift mutation: (completely absent of β- globin)
3. Mutation in regulatory sequences (promotor and two enhancers): result in reduced
synthesis of mRNA and a reduction, but not absence of β- globin.
4. Splice site consensus mutations: produce less sever disease.