Etiology:
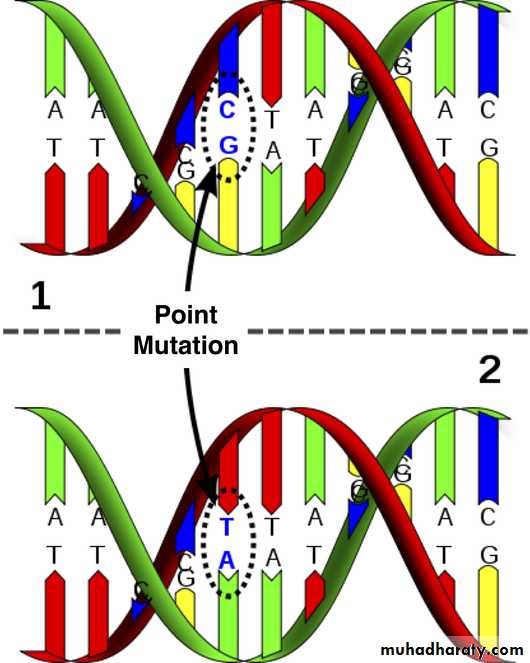
Mutation refers to permanent changes in the DNA. Those that affect germ cells are transmitted to the progeny and may give rise to inherited diseases. Mutations in somatic cells are not transmitted to the progeny but are important in the causation of cancers and some congenital malformationsThus there is certain changes occur in nitrogenous bases may lead to abnormal protein synthesis.
All single gene diseases are due to mutations. They are of different types:
Single point mutation which is the commonest. They usually result from a change in one of the nucleotide bases that form the trios (three bases), each of which codes for a specific amino acid in the protein molecule. Not all of these changes will result in a mutation that causes a disease.
2-Additional – deletional mutations : They could be one of three types :
Addition or deletion of a single base.Addition or deletion of 3 bases or the multiple of 3,
Addition or deletion of a large piece of DNA inside the gene (intragenic) or in between the genes (intergenic). Again this creates variability and it is used for genetic testing and diagnosis of genetic diseases.

Chromosomal Diseases
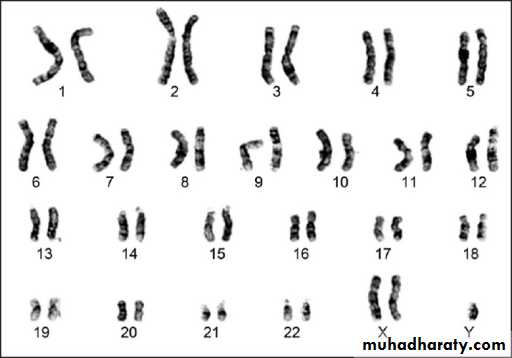
These are classified into:a. Numerical abnormalities, which is defined as a gain or loss of a whole chromosome from the usual number of chromosomes in the karyotype (i.e. 46 chromosomes). Gain or loss in the sex chromosomes especially the X-chromosome is compatible with life and is relatively common; while loss of an autosomal chromosome is usually non-viable and a fertilized ovum carrying such karyotype could not sustain pregnancy to full term and usually are lost very early in pregnancy, i.e. abortion.
Autosomal chromosome trisomy is exemplified by trisomy 21 or Down’s syndrome, trisomy 18 or Edward’s syndrome, trisomy 13 or Patau’s syndrome.
Sex chromosome trisomy is exemplified by Klienfilter’s syndrome in male and triple X (XXX) syndrome in female or XYY syndrome in male, while monosomy of sex chromosome is when a female loses one X resulting in Turner’s syndrome, 45 X known as aneuploidy.
b. Structural abnormalities: it usually results from breakage followed by rearrangement of material (a cell suffering from structural abnormality has the normal number of 46 but the chromosomes are morphologically or structurally abnormal).
These abnormalities are of different types:
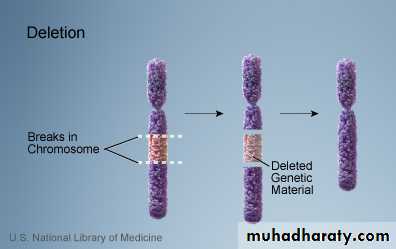
A. Deletion: loss of a piece of a chromosome. It is of two types
i. Terminal: single break may delete a terminal segment.
ii. interstitial: where the piece of a chromosome between two breaks is lost resulting in a syndrome
B. Inversion:
This abnormality results from two breaks through out the length of the chromosome which either involve the centromere area or not and the piece between the two breaks will rotate 180o before it returns to its place. So the genetic piece which is broken it will rotated so the position of the gene occupied by this segment is abnormal & the content of the genetic material are changed according to the piece which is inverted & this will result in abnormal fetus with signs & symptoms of abnormality.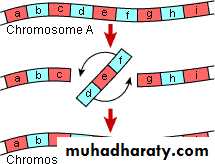
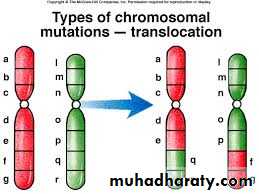
C.Translocation: This is defined as exchange of segments of chromosomes between two non-homologous chromosomes so there is a translocation of some oncogenes from their normal habitat to a new situation where they are induced to function in uncontrolled manner leading to malignancies. This is usually seen in leukemias and lymphomas, e.g. Philadelphia chromosome.
D. Isochromosome: This abnormality results from aberrant division of the centromere which is the last part of the chromosome that divides in the mitosis to separate the two sister chromatids into individual chromosomes. This aberrant division takes place in a horizontal way rather than the perpendicular natural way. So the resulting two chromosomes are imbalanced, one formed of two short arms and the other of two long arms. Each of them is an isochromsome.

E. Ring-chromosome: It results from deletion of both ends of a chromosome and then the ends, because of the adhesive nature of the exposed DNA, will stick together forming a ring.
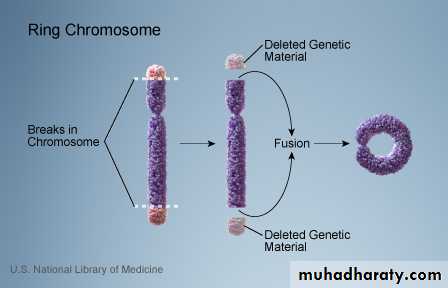
Cytogenetic disorders involving autosomes:
Down syndrome (Trisomy 21), karytype (47, xx or xy, +21):Is the most common of the chromosomal disorders. About 95% of affected persons have trisomy 21 resulting from meiotic nondisjunction. The parents of such children have normal karyotype and are normal in all respects. Increasing of maternal age has a strong influence on the incidence of Down syndrome. The correlation with maternal age suggests that in most cases the meiotic nondisjunction of chromosome 21 occurs in the ovum.
In about 4% of all patients with trisomy 21, the extra chromosomal material is due to translocation of the long arm chromosome 21 to chromosome 22 or 14 and the remaining 1% of trisomy 21 patients are mosaics.
Trisomy 21 is the leading cause of mental retardation. The patient has a combination of epicanthic folds and flat facial profile which is quite characteristic. Congenital malformations are common. Approximately 40% of patients have cardiac malformations, which are responsible for most of the death in early childhood. They found that approximately 80% of those without congenital heart disease can expect to survive 30 years but most of them develop Alzheimer disease and frank dementia.
Serious infections and increased risk of developing acute leukemias are another cause of morbidity and mortality in patients with trisomy 21.

Cytogenetic disorders involving sex chromosome:
Klinefelter syndrome: this syndrome is best defined as male hypogonadism that develop when there are at least two X chromosomes and one or more Y chromosomes. Most patients are 47, XXY. This karyotype results from nondisjunction of sex chromosomes during meiosis. The extra X chromosome may be of maternal or paternal origin. Advanced maternal age and history of irradiation of either parent may contribute to the meiotic error resulting in this condition. Approximately 15% of patients show mosaic patterns (i.e. 46,XY/47,XXY or 47,XXY/48,XXXY) and the presence of a 46,XY line in mosaics is usually associated with a milder clinical condition.Although the following description applies to most patients, it should be noted that klinefelter syndrome is associated with a wide rang of clinical manifestations. In some it may be expressed only as hypogonadism, but most patients have an increased length between the soles and the pubic bone, which creates the appearance of an elongated body. Reduced facial body and pubic hair with gyneocomastia are also frequently noted. Testicular atrophy, the serum testosterone levels are lower than normal while urinary gonadotropin levels are elevated. So the principle clinical effect of this syndrome is sterility. Only rarely the patients are fertile and these are presumably mosaics with a large proportion of 46, XY cells. This syndrome may be associated with mental retardation but the degree of intellectual impairment is typically mild and in some cases is undetectable. The reduction in intelligence is correlated with the number of extra X chromosomes. Thus, in patients with the most common variant (XXY), intelligence is nearly normal, but in those with rare variant forms involving additional X chromosomes, significantly subnormal levels of intelligence, as well as more sever physical abnormalities are found.
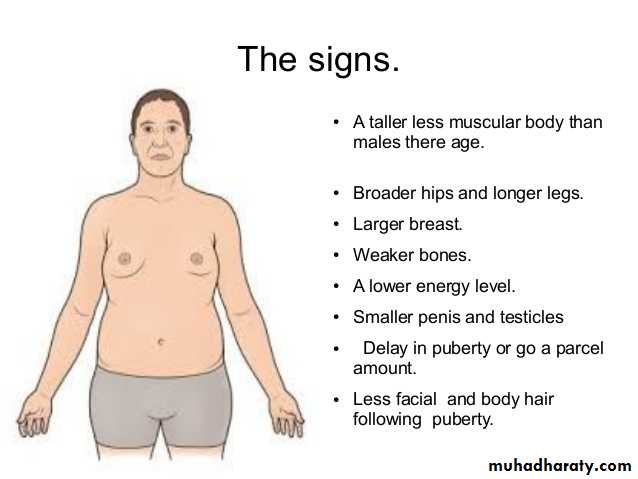
Turner syndrome:
It is characterized by primary hypogonadism in phenotypic females, results from partial or complete monosomy of the short arm of the X chromosome. In approximately 57% of patients, the entire X chromosome is missing, resulting in a 45,X karyotype. These patients are the most severely affected. Typical clinical features associated with 45X Turner syndrome include significant growth retardation, leading to abnormal short stature; swelling of the nap of the neck due to distended lymphatic channels (in infancy) that is seen as webbing of the neck in older children; low posterior hair line; shieldlike chest with widely spaced nipples; high arched palate; lymphedema of the hands and feet; and a variety of congenital malformations such as horseshoe kidney, bicuspid aortic valve and coarctation of the aorta. Affected females develop normal secondary sex characteristics; the genitalia remains infantile, breast development is minimal and little pubic hair appears. Most have primary amenorrhea, and morphologic examination reveals transformation of the ovaries into white streaks of fibrous stroma devoid of follicles. The mental status of these patients is usually normal. Curiously, hypothyroidism caused by autoantibodies is noted in 25% to 30%. In adult patients a combination of short stature and primary amenorrhea should prompt strong suspicion of Turner syndrome. The diagnosis is established by karyotyping.Approximately 43% of patients with turner syndrome either are mosaics (one of the cell lines being 45,X) or have structural abnormalities of the X chromosome.