
Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine
2016

learning-topics
Endocrine diseaseEndocrinology concerns the synthesis, secretion and action of hormones. These are chemical messengers released from endocrine glands that coordinate the activities of many different cells. Endocrine diseases can therefore affect multiple organs and systems.
Endocrine disease causes clinical syndromes with symptoms and signs involving many organ systems, reflecting the diverse effects of hormone deficiency and excess. The
emphasis of the clinical examination depends on the gland or hormone that is thought to be abnormal.
Few endocrine therapies have been evaluated by randomized controlled trials, in part because hormone
replacement therapy (for example, with levothyroxine)
has obvious clinical benefits and placebo-controlled
trials would be unethical, and in part because many
endocrine diseases are rare, making trials difficult to
perform. Recommendations for ‘evidence-based medicine’ are, therefore, relatively scarce. They relate mainly to use of therapy that is ‘optional’ and/or recently available, such as oestrogen replacement in post-menopausal women, androgen therapy in older men and growth hormone replacement.
AN OVERVIEW OF ENDOCRINOLOGY
Functional anatomy and physiology
Some endocrine glands, such as the parathyroids and
pancreas, respond directly to metabolic signals, but most
are controlled by hormones released from the pituitary
gland.
Anterior pituitary hormone secretion is controlled
in turn by substances produced in the hypothalamus
and released into portal blood, which drains directly
down the pituitary stalk .
Posterior pituitary hormones are synthesised in the hypothalamus and transported down nerve axons, to be released from the posterior pituitary.
Hormone release in the hypothalamus and pituitary is regulated by numerous stimuli and through feedback control by hormones produced by the target glands (thyroid, adrenal cortex and gonads).
These integrated endocrine systems are called ‘axes’.
The biological effects of most hormones are mediated by binding to receptors on the cell surface. These interact with various intracellular signalling molecules on the cytosolic side of the plasma membrane to affect cell function, usually through changes in gene expression . Some hormones, most notably steroids, triiodothyronine and vitamin D, bind to specific intracellular receptors, which directly bind to response elements on DNA to regulate gene expression.
The classical model of endocrine function involves hormones synthesised in endocrine glands, which are released into the circulation and act at sites distant from those of secretion . However, additional levels of regulation are now recognised. Many other organs secrete hormones or contribute to the peripheral metabolism and activation of pro-hormones. A notable example is the production of oestrogens from adrenal androgens in adipose tissue by the enzyme aromatase. Some hormones, such as neurotransmitters, act in a paracrine fashion to affect adjacent cells, or act in an autocrine way to affect behaviour of the cell that produces the hormone.
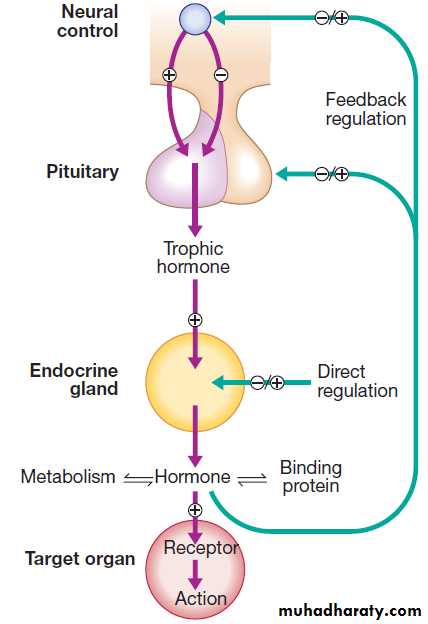
An archetypal endocrine axis. Regulation by negative
feedback and direct control is shown, along with the equilibrium between active circulating free hormone and bound or metabolised hormone.
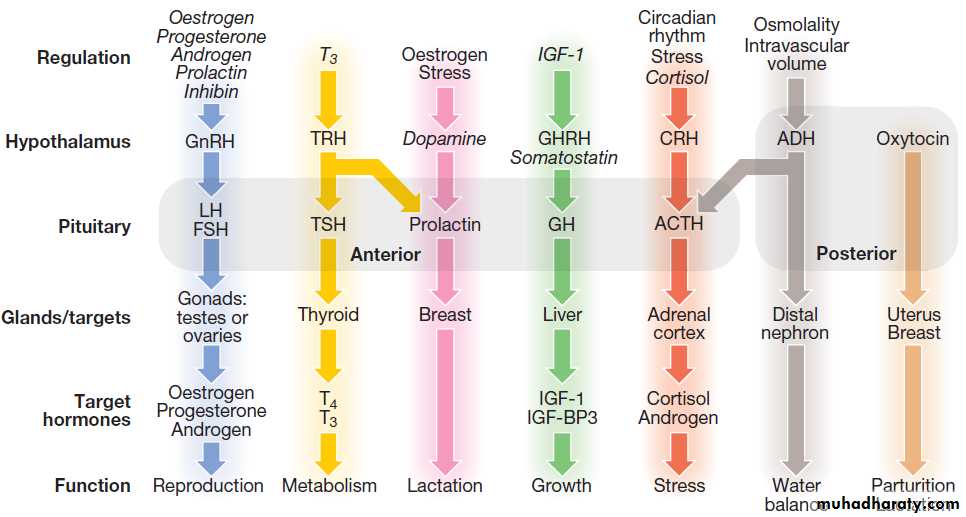
Fig.The principal endocrine ‘axes’. Some major endocrine glands are not controlled by the pituitary. These include the parathyroid glands (regulated by calcium concentrations), the adrenal zona glomerulosa (regulated by the renin–angiotensin system) and the endocrine pancreas . Italics show negative regulation. (ACTH = adrenocorticotrophic hormone; ADH = antidiuretic hormone, arginine vasopressin; CRH = corticotrophin-releasing hormone; FSH = follicle-stimulating hormone; GH = growth hormone; GHRH = growth hormone-releasing hormone; GnRH = gonadotrophin-releasing hormone; IGF-1 = insulin-like growth factor-1; IGF-BP3 = IGF-binding protein-3; LH = luteinising hormone:
T3 = triiodothyronine; T4 = thyroxine; TRH = thyrotrophin-releasing hormone; TSH = thyroid-stimulating hormone).
Endocrine pathology
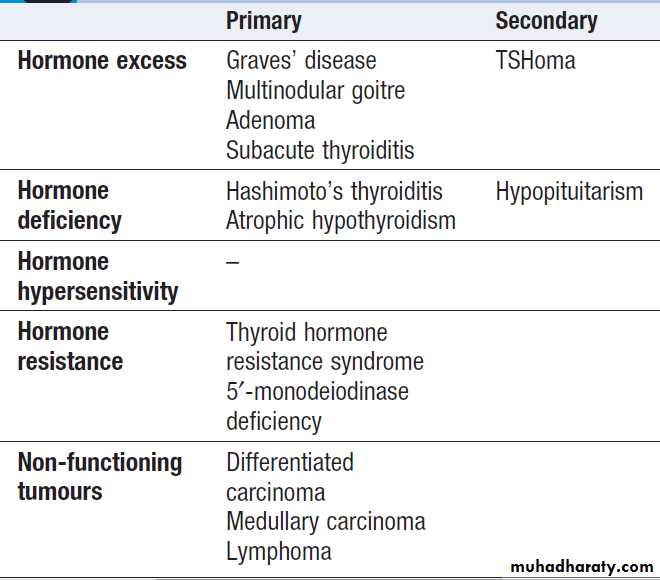
For each endocrine axis or major gland, diseases can beclassified as shown in Box. Pathology arising within
the gland is often called ‘primary’ disease (for example,
primary hypothyroidism in Hashimoto’s thyroiditis),
while abnormal stimulation of the gland is often called
‘secondary’ disease (for example, secondary hypothyroidism in patients with a pituitary tumour and
thyroid-stimulating hormone deficiency). Some pathological
processes can affect multiple endocrine glands, these may have a genetic basis (such as organspecific autoimmune endocrine disorders and the multiple endocrine neoplasia (MEN) syndromes) or be a consequence of therapy for another disease (for example, following treatment of childhood cancer with chemotherapy and/or radiotherapy).

Classification of endocrine disease
Investigation of endocrine diseaseBiochemical investigations play a central role in
endocrinology. Most hormones can be measured in blood, but the circumstances in which the sample is taken are often crucial, especially for hormones with pulsatile secretion, such as growth hormone; those that show diurnal variation, such as cortisol; or those that demonstrate monthly variation, such as oestrogen or progesterone.
Other investigations, such as imaging and biopsy, are more frequently reserved for patients who present with a tumour. The principles of investigation are shown in Box.
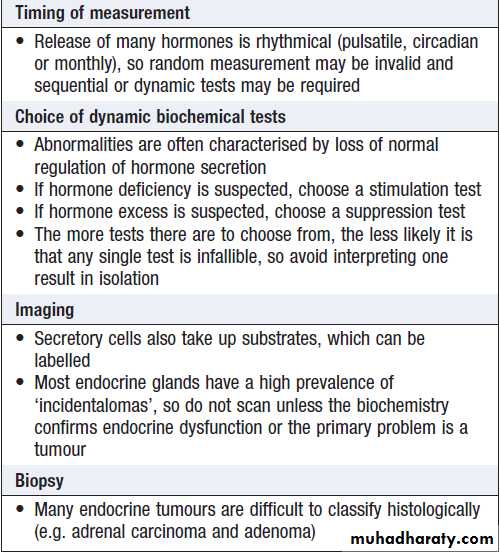
Principles of endocrine investigation
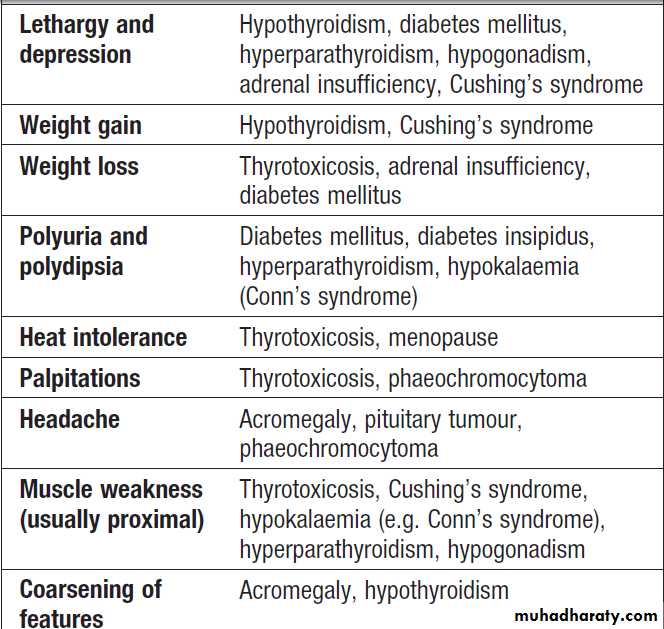

Examples of non-specific presentations of endocrine disease
Classification of thyroid disease
THE THYROID GLANDDiseases of the thyroid predominantly affect females
and are common, occurring in about 5% of the population Functional anatomy, physiology and investigations
The parafollicular C cells secrete calcitonin, which is of
no apparent physiological significance in humans. The
follicular epithelial cells synthesise thyroid hormones
by incorporating iodine into the amino acid tyrosine on
the surface of thyroglobulin (Tg), a protein secreted into
the colloid of the follicle.
Iodide is a key substrate for thyroid hormone synthesis; a dietary intake in excess of 100 μg/day is required to maintain thyroid function in adults.
The thyroid secretes predominantly thyroxine (T4) and only a small amount of triiodothyronine (T3); approximately 85% of T3 in blood is produced from T4 by a family of monodeiodinase enzymes which are active in many tissues, including liver, muscle, heart and kidney. Selenium is an integral component of these monodeiodinases. T4 can be regarded as a pro-hormone, since it has a longer half-life in blood than T3 (approximately 1 week compared with approximately 18 hours), and binds and activates thyroid hormone receptors less effectively than T3. T4 can also be converted to the inactive metabolite, reverse T3.
T3 and T4 circulate in plasma almost entirely (> 99%)
bound to transport proteins, mainly thyroxine-bindingglobulin (TBG). It is the unbound or free hormones
which diffuse into tissues and exert diverse metabolic
actions. Some laboratories use assays which measure
total T4 and T3 in plasma, but it is increasingly common
to measure free T4 and free T3. The advantage of the free hormone measurements is that they are not influenced by changes in the concentration of binding proteins; in pregnancy, for example, TBG levels are increased and total T3 and T4 may be raised, but free thyroid hormone levels are normal.
Production of T3 and T4 in the thyroid is stimulated by thyrotrophin (TSH), a glycoprotein released from the thyrotroph cells of the anterior pituitary in response to the hypothalamic tripeptide, TRH.
A circadian rhythm of TSH secretion can be demonstrated
with a peak at 0100 hrs and trough at 1100 hrs, but the
variation is small so that thyroid function can be assessed
reliably from a single blood sample taken at any time of
day and does not usually require any dynamic stimulation
or suppression tests. There is a negative feedback of
thyroid hormones on the hypothalamus and pituitary
such that in thyrotoxicosis, when plasma concentrations
of T3 and T4 are raised, TSH secretion is suppressed.
Conversely, in hypothyroidism due to disease of the
thyroid gland, low T3 and T4 are associated with high circulating TSH levels.The anterior pituitary is very sensitive to minor changes in thyroid hormone levels within the reference range.
For this reason, TSH is usually regarded as the most useful investigation of thyroid function.
However, interpretation of TSH values without considering
thyroid hormone levels may be misleading in patients with pituitary disease .
Moreover, TSH may take several weeks to ‘catch up’
with T4 and T3 levels, for example, when prolongedsuppression of TSH in thyrotoxicosis is relieved by
Antithyroid therapy. Heterophilic antibodies can also
interfere with the TSH assay and cause a spuriously high
measurement.
Other modalities commonly employed in the investigation
of thyroid disease include measurement of antibodies
against the TSH receptor or other thyroid antigens,
radioisotope imaging, fine needle aspiration biopsy and ultrasound. Their use is described below.
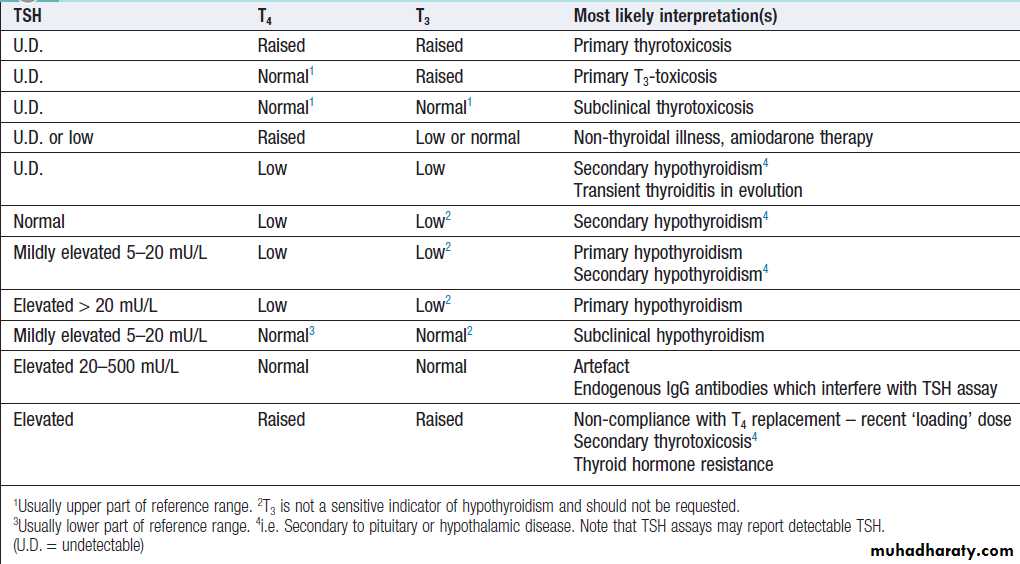
How to interpret thyroid function test results
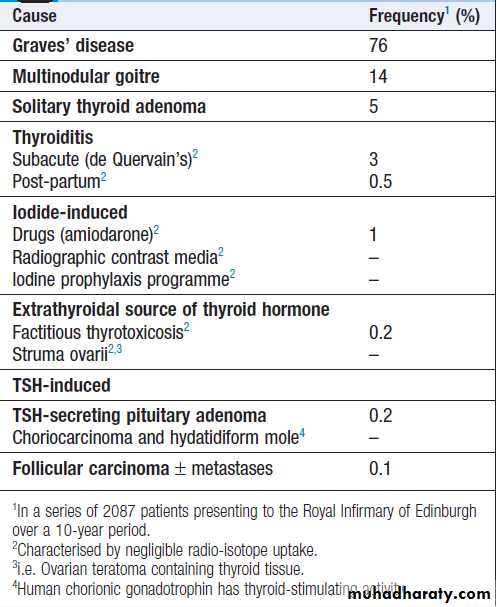
Causes of thyrotoxicosis and their
relative frequenciesThyrotoxicosis
Thyrotoxicosis describes a constellation of clinical featuresarising from elevated circulating levels of thyroid
hormone. The most common causes are Graves’ disease,
multinodular goitre and autonomously functioning
thyroid nodules (toxic adenoma) . Thyroiditis
is more common in parts of the world where relevant
viral infections occur, such as North America.
Clinical assessment
The most common symptoms are weight loss with a normal or increased appetite, heat intolerance, palpitations, tremor and irritability. Tachycardia, palmar erythema and lid lag are common signs.
Not all patients have a palpable goitre, but experienced clinicians can discriminate the diffuse soft goitre of Graves’ disease from the irregular enlargement
of a multinodular goitre. All causes of thyrotoxicosis can cause lid retraction and lid lag, due to potentiation
of sympathetic innervation of the levator palpebrae
muscles, but only Graves’ disease causes other features
of ophthalmopathy, including periorbital oedema, conjunctival irritation, exophthalmos and diplopia. Pretibial myxoedema and the rare thyroid acropachy (a
periosteal hypertrophy, indistinguishable from finger
clubbing) are also specific to Graves’ disease.
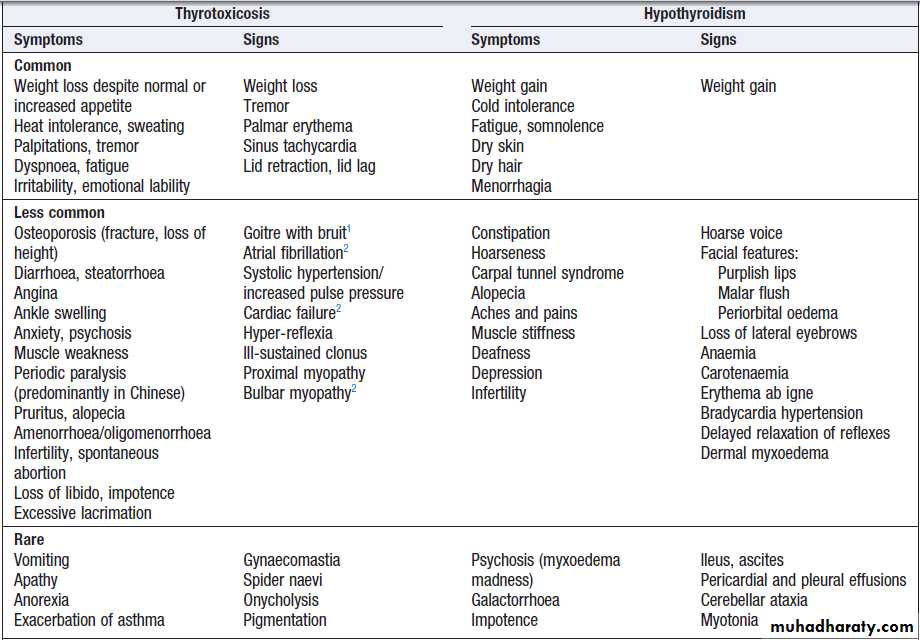

Clinical features of thyroid dysfunction
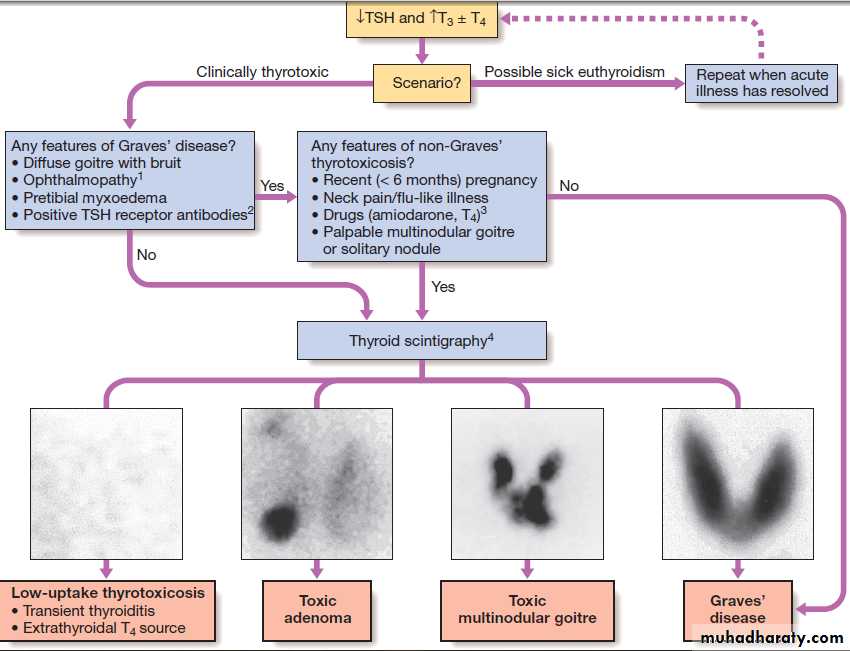
Fig. Establishing the differential diagnosis in thyrotoxicosis. (1) Graves’ ophthalmopathy refers to clinical features of exophthalmos and periorbital and conjunctival oedema, not simply the lid lag and lid retraction which can occur in all forms of thyrotoxicosis. (2) TSH receptor antibodies are very rare in patients without autoimmune thyroid disease, but only occur in 80–95% of patients with Graves’ disease; a positive test is therefore confirmatory, but a negative test does not exclude Graves’ disease. Other thyroid antibodies (e.g. anti-peroxidase and anti-thyroglobulin antibodies) are unhelpful in the differential diagnosis since they occur frequently in the population and are found with several of the disorders which cause thyrotoxicosis.
(3) Scintigraphy is not necessary in most cases of drug-induced thyrotoxicosis.
(4) 99mTechnetium pertechnetate scans of patients with thyrotoxicosis. In low-uptake thyrotoxicosis, most commonly due to a viral, post-partum or iodine-induced thyroiditis, there is negligible isotope detected in the region of the thyroid, although uptake is apparent in nearby salivary glands (not shown here). In a toxic adenoma there is lack of uptake of isotope by the rest of the thyroid gland due to suppression of serum TSH. In multinodular goitre there is relatively low, patchy uptake within the nodules; such an appearance is not always associated with a palpable thyroid. In Graves’ disease there is diffuse uptake of isotope.
Investigations
The first-line investigations are serum T3, T4 and TSH. If
abnormal values are found, the tests should be repeated
and the abnormality confirmed in view of the likely need
for prolonged medical treatment or destructive therapy.
In most patients, serum T3 and T4 are elevated, but in about 5% T4 is in the upper part of the reference range and T3 raised (T3 toxicosis).
Serum TSH is undetectable in primary thyrotoxicosis, but values can be raised in the very rare syndrome of secondary thyrotoxicosis caused by a TSH-producing pituitary adenoma.
When biochemical thyrotoxicosis has been confirmed, further investigations should be undertaken to determine the underlying cause, including measurement of TSH receptor antibodies (TRAb, elevated in Graves’ disease) and isotope scanning.
An ECG may demonstrate sinus tachycardia or atrial
fibrillation.
Radio-iodine uptake tests measure the proportion
of isotope that is trapped in the whole gland, but have
been largely superseded by 99mtechnetium scintigraphy
scans, which also indicate trapping, are quicker to
perform with a lower dose of radioactivity, and provide
a higher-resolution image. In low-uptake thyrotoxicosis,
the cause is usually a transient thyroiditis .
Occasionally, patients induce ‘factitious thyrotoxicosis’ by
consuming excessive amounts of a thyroid hormonepreparation, most often levothyroxine.
The exogenous thyroxine suppresses pituitary TSH secretion and hence iodine uptake, serum thyroglobulin and release of endogenous thyroid hormones. The T4:T3 ratio (typically
30 : 1 in conventional thyrotoxicosis) is increased to
above 70 : 1 because circulating T3 in factitious thyrotoxicosis is derived exclusively from the peripheral monodeiodination of T4 and not from thyroid secretion. The
combination of negligible iodine uptake, high T4:T3 ratio
and a low or undetectable thyroglobulin is diagnostic.
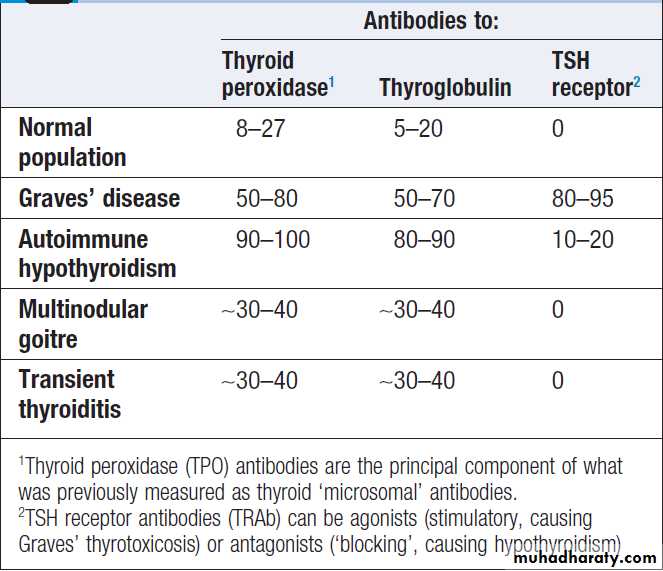
Prevalence of thyroid autoantibodies (%)
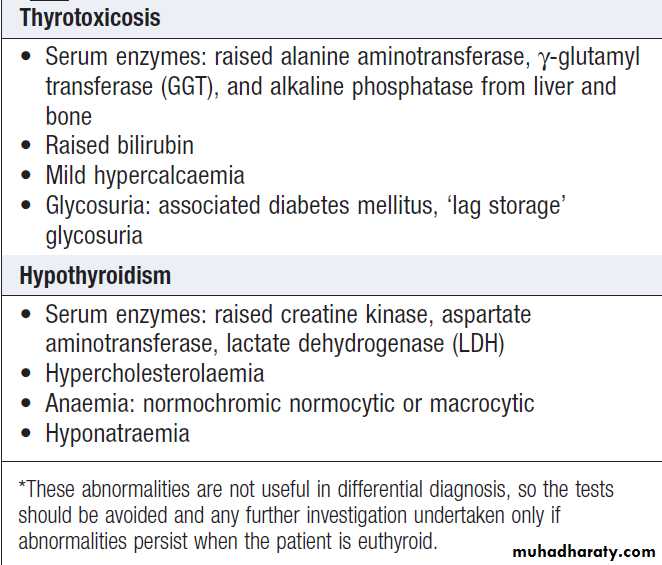
Non-specific laboratory abnormalities in
thyroid dysfunction*Management
Definitive treatment of thyrotoxicosis depends onthe underlying cause and may include antithyroid
drugs, radioactive iodine or surgery. A non-selective β- adrenoceptors antagonist (β-blocker), such as propranolol (160 mg daily) or nadolol (40–80 mg daily), will alleviate
but not abolish symptoms in most patients within 24–48
hours. Beta-blockers should not be used for long-term
treatment of thyrotoxicosis but are extremely useful in
the short term, whilst patients are awaiting hospital consultation or following 131I therapy.
Atrial fibrillation in thyrotoxicosis
Atrial fibrillation occurs in about 10% of patients withthyrotoxicosis. The incidence increases with age, so that
almost half of all males with thyrotoxicosis over the age
of 60 are affected. Moreover, subclinical thyrotoxicosis
is a risk factor for atrial fibrillation. Characteristically,
the ventricular rate is little influenced by digoxin, but responds to the addition of a β-blocker. Thromboembolic vascular complications are particularly common in thyrotoxic atrial fibrillation so that anticoagulation with warfarin is required, unless contraindicated. Once thyroid hormone and TSH have been returned to normal, atrial fibrillation will spontaneously revert to sinus rhythm in about 50%, but cardioversion may be required in the remainder.
Thyrotoxic crisis (‘thyroid storm’)
This is a rare but life-threatening complication of thyrotoxicosis. The most prominent signs are fever, agitation, confusion, tachycardia or atrial fibrillation and, in the older patient, cardiac failure. It is a medical emergency, which has a mortality of 10% despite early recognition and treatment. Thyrotoxic crisis is most commonly precipitated by infection in a patient with previously unrecognized or inadequately treated thyrotoxicosis. It may
also develop shortly after subtotal thyroidectomy in an
ill-prepared patient or within a few days of 131I therapy,
when acute irradiation damage may lead to a transient
rise in serum thyroid hormone levels.
Patients should be rehydrated and given propranolol, either orally (80 mg 4 times daily) or IV (1–5 mg 4 times daily).
Sodium ipodate (500 mg per day orally) will restore serum T3 to normal in 48–72 hours. This is a radiographic contrast medium which not only inhibits the release of thyroid hormones, but also reduces the conversion of T4 to T3 and is, therefore, more effective than potassium iodide or Lugol’s solution. Dexamethasone (2 mg 4 times daily) and amiodarone have similar effects. Oral carbimazole 40–60 mg daily should be given to inhibit the synthesis of new thyroid hormone. If the patient is unconscious or
uncooperative, carbimazole can be administered rectally
with good effect, but no preparation is available for
parenteral use. After 10–14 days the patient can usually
be maintained on carbimazole alone.
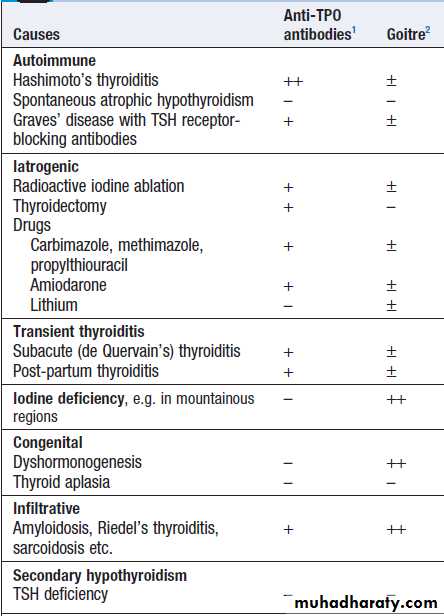
Causes of hypothyroidism
1thyroid autoantibodies are common in the health population, so might be present in anyone. ++ high titre; + more likely to be detected than in the healthy population; – not especially likely.2Goitre: – absent; ± may be present; ++ characteristic.
Hypothyroidism
Hypothyroidism is a common condition with various
causes , but autoimmune disease (Hashimoto’s thyroiditis) and thyroid failure following 131I or surgical treatment of thyrotoxicosis account for over 90% of cases, except in areas where iodine deficiency is endemic.
Women are affected approximately six times more frequently than men.
Clinical assessment
The clinical presentation depends on the duration and
severity of the hypothyroidism. Those in whom complete
thyroid failure has developed insidiously over months or years may present with many of the clinical features listed.
A consequence of prolonged hypothyroidism is the infiltration of many body tissues by the mucopolysaccharides, hyaluronic acid and chondroitin
sulphate, resulting in a low-pitched voice, poor hearing, slurred speech due to a large tongue. Compression
of the median nerve at the wrist (carpal tunnel syndrome). Infiltration of the dermis gives rise to nonpitting oedema (myxoedema), which is most marked in the skin of the hands, feet and eyelids. The resultant periorbital puffiness is often striking and may be combined with facial pallor due to vasoconstriction and
anaemia, or a lemon-yellow tint to the skin caused by
carotenaemia, along with purplish lips and malar flush.
Most cases of hypothyroidism are not clinically obvious,
however, and a high index of suspicion needs to bemaintained so that the diagnosis is not overlooked in
individuals complaining of non-specific symptoms such
as tiredness, weight gain, depression or carpal tunnel
syndrome. Care must be taken to identify patients with transient hypothyroidism, in whom life-long levothyroxine therapy is inappropriate. This is often observed during the first 6 months after subtotal thyroidectomy or 131I treatment of Graves’ disease, in the post-thyrotoxic phase of subacute thyroiditis and in post-partum thyroiditis. In these conditions, levothyroxine treatment is not always necessary, as the patient may be asymptomatic during the short period of thyroid failure.
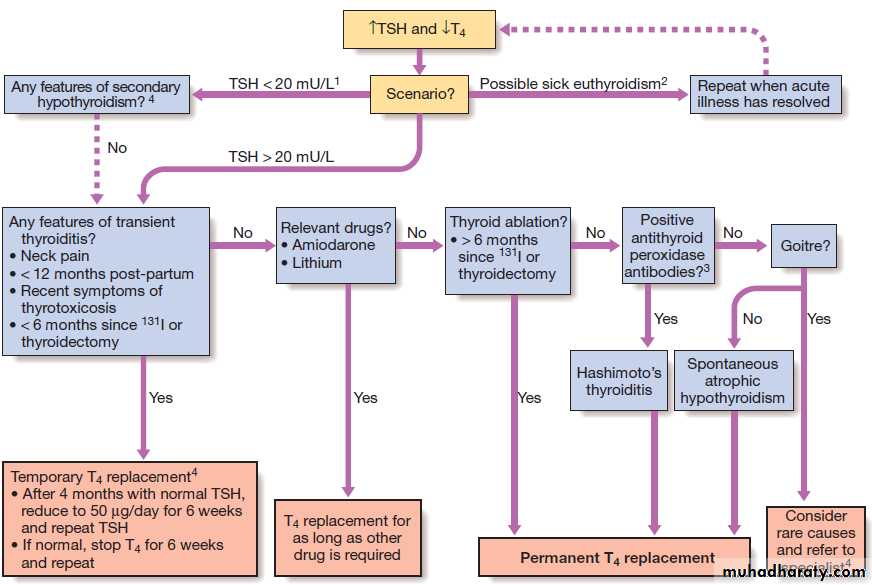
Fig. An approach to adults with suspected primary hypothyroidism. This scheme ignores congenital causes of hypothyroidism such as thyroid aplasia and dyshormonogenesis (associated with nerve deafness in Pendred’s syndrome), which are usually diagnosed in
childhood. (1) Immunoreactive TSH may be detected at normal or even modestly elevated levels in patients with pituitary failure; unless T4 is only marginally low, TSH should be > 20 mU/L to confirm the diagnosis of primary hypothyroidism. (2) The usual abnormality in sick euthyroidism is a low TSH but any pattern can occur. (3) Thyroid peroxidase antibodies are highly sensitive but not very specific for autoimmune thyroid disease . (4) Specialist advice is most appropriate where indicated. Secondary hypothyroidism is rare, but is suggested by deficiency of pituitary hormones or by clinical features of pituitary tumour such as headache or visual field defect .
Investigations
In the vast majority of cases, hypothyroidism results from an intrinsic disorder of the thyroid gland (primaryhypothyroidism). In this situation, serum T4 is low and TSH is elevated, usually in excess of 20 mU/L. Measurements of serum T3 are unhelpful since they do not discriminate reliably between euthyroidism and hypothyroidism. Secondary hypothyroidism is rare and is caused by failure of TSH secretion in an individual with hypothalamic or anterior pituitary disease. In severe, prolonged hypothyroidism, the ECG classically demonstrates sinus bradycardia with low-voltage complexes and ST segment and T-wave abnormalities. Measurement of thyroid peroxidase antibodies is helpful but further investigations are rarely required .
Management
Treatment is with levothyroxine replacement. It is customaryto start with a low dose of 50 μg per day for 3 weeks, increasing thereafter to 100 μg per day for a further 3 weeks and finally to a maintenance dose of 100–150 μg per day. In younger patients, it is safe to initiate levothyroxine at a higher dose (for example, 100 μg per day), to allow a more rapid normalisation of thyroid hormone levels. Levothyroxine has a half-life of
7 days so it should always be taken as a single daily dose
and at least 6 weeks should pass before repeating
thyroid function tests and adjusting the dose, usually
by 25 μg per day. Patients feel better within 2–3 weeks.
Reduction in weight and periorbital puffiness occurs
quickly, but the restoration of skin and hair texture andresolution of any effusions may take 3–6 months. As
illustrated in Figure, most patients require life-long levothyroxine therapy. The dose should be adjusted to maintain serum TSH within the reference range. To achieve this, serum T4 often needs to be in the upper part of the reference range or even slightly raised, because the T3 required for receptor activation is derived exclusively from conversion of T4 within the target tissues, without the usual contribution from thyroid secretion. Some physicians advocate combined replacement with T4 and T3 or preparations of animal thyroid extract, but this approach remains controversial.
Some patients remain symptomatic despite normalisation of TSH and may wish to take extra levothyroxine, which suppresses TSH. However, suppressed TSH is a risk factor for osteoporosis and atrial fibrillation (subclinical thyrotoxicosis), so this approach cannot be recommended.
It is important to measure thyroid function every
1–2 years once the dose of levothyroxine is stabilised.
This encourages patient compliance with therapy and
allows adjustment for variable underlying thyroid activity
and other changes in levothyroxine requirements.
Some patients have a persistent elevation of serum TSH despite an ostensibly adequate replacement dose of levothyroxine; most commonly, this is a consequence
of suboptimal compliance with therapy.
There may be differences in bioavailability between the numerous generic preparations of levothyroxine and so, if an individual is experiencing marked changes in serum
TSH despite optimal compliance, the prescription of a
branded preparation of levothyroxine could be considered.
Levothyroxine absorption is maximal when the
medication is taken before bed and may be further optimized by taking a vitamin C supplement.
In some poorly compliant patients, levothyroxine is
taken diligently or even in excess for a few days prior
to a clinic visit, resulting in the seemingly anomalous
combination of a high serum T4 and high TSH .
Levothyroxine replacement in ischaemic heart disease
Although angina may remain unchanged in severity or paradoxically disappear with restoration of metabolic rate, exacerbation of myocardial ischaemia, infarction and sudden death are recognised complications of levothyroxine replacement, even using doses as low as 25 μg per day. In patients with known IHD, thyroid replacement should be introduced at low dose and increased very slowly under specialist supervision. It has been suggested that T3 has an advantage over T4, since T3 has a shorter half-life and any adverse effect will reverse more quickly, but the more distinct peak in hormone levels after each dose of T3 is a disadvantage.Coronary angioplasty or bypass surgery may be required if angina is exacerbated by levothyroxine therapy.
Hypothyroidism in pregnancy
Most pregnant women with primary hypothyroidism require an increase in the dose of levothyroxine of approximately 25–50 μg daily to maintain normal TSH levels. This may reflect increased metabolism of thyroxine by the placenta and increased serum thyroxine binding globulin during pregnancy, resulting in an increase in the total thyroid hormone pool to maintain the same free T4 and T3 concentrations. Inadequate maternal T4 therapy may be associated with impaired cognitive development in an unborn child and so women are usually advised to increase their daily levothyroxine dose by 25 μg when pregnancy is confirmed. Serum TSH and free T4 should be measured during each trimester and the dose of levothyroxine adjusted to maintain a normal TSH.
Myxoedema coma
This is a very rare presentation of hypothyroidism inwhich there is a depressed level of consciousness,
usually in an elderly patient who appears myxoedematous.
Body temperature may be as low as 25°C, convulsions
are not uncommon and cerebrospinal fluid (CSF) pressure and protein content are raised.
The mortality rate is 50% and survival depends on early recognition and treatment of hypothyroidism and other factors contributing to the altered consciousness level, such as medication, cardiac failure, pneumonia, dilutional hyponatraemia and respiratory failure.
Myxoedema coma is a medical emergency and treatment
must begin before biochemical confirmation of thediagnosis. Suspected cases should be treated with an
intravenous injection of 20 μg triiodothyronine, followed
by further injections of 20 μg 3 times daily until
there is sustained clinical improvement. In survivors,
there is a rise in body temperature within 24 hours and,
after 48–72 hours, it is usually possible to switch patients
to oral levothyroxine in a dose of 50 μg daily.
Unless it is apparent that the patient has primary hypothyroidism, the thyroid failure should also be assumed to be secondary to hypothalamic or pituitary disease and treatment given with hydrocortisone 100 mg IM 3 times daily, pending the results of T4, TSH and cortisol
measurement . Other measures include slow
rewarming , cautious use of intravenous fluids,
broad-spectrum antibiotics and high-flow oxygen. Occasionally, assisted ventilation may be necessary.
Symptoms of hypothyroidism with normal
thyroid function testsThe classic symptoms of hypothyroidism are, by their very nature, non-specific . There is a wide differential diagnosis for symptoms such as ‘fatigue’, ‘weight gain’ and ‘low mood’. Serum TSH is an excellent measure of an individual’s thyroid hormone status. However, some individuals believe that they have hypothyroidism despite normal serum TSH concentrations. There are a large number of websites which claim that serum TSH is not a good measure of thyroid hormone status and suggest that other factors, such as abnormalities of T4 to T3 conversion, may lead to low tissue levels of active thyroid hormones.
Such websites often advocate a variety of tests of thyroid function of dubious scientific validity, including measurement of serum reverse T3, 24-hour urine T3, basal body temperature, skin iodine absorption, and levels of selenium in blood and urine. Individuals who believe they have hypothyroidism, despite normal conventional tests of thyroid function, can be difficult to manage.
They require reassurance that their symptoms are being taken seriously and that organic disease has been carefully considered; if their symptoms persist, then referral to a team specialising in medically unexplained symptoms should be considered.
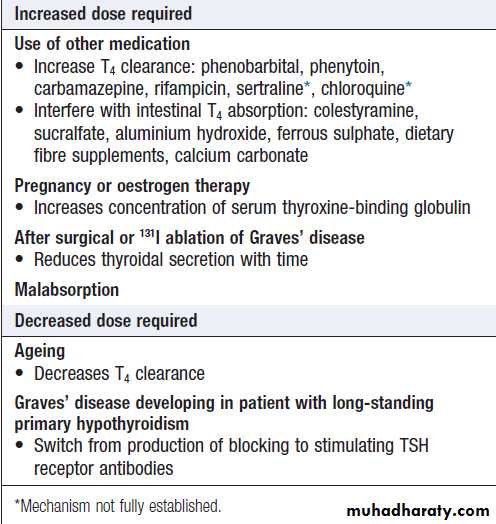
Situations in which an adjustment of
the dose of levothyroxine may be necessaryAsymptomatic abnormal thyroid function tests
One of the most common problems in medical practiceis how to manage patients with abnormal thyroid function tests who have no obvious signs or symptoms of thyroid disease. These can be divided into three categories.
Subclinical thyrotoxicosis
Serum TSH is undetectable, and serum T3 and T4 are at the upper end of the reference range. This combination is most often found in older patients with multinodular goitre. These patients are at increased risk of atrial fibrillation and osteoporosis, and hence the consensus view is that they have mild thyrotoxicosis and require therapy, usually with 131I. Otherwise, annual review is essential, as the conversion rate to overt thyrotoxicosis with elevated T4 and/or T3 concentrations is 5% each year.
Subclinical hypothyroidism
Serum TSH is raised, and serum T3 and T4 concentrations
are at the lower end of the reference range. This may
persist for many years, although there is a risk of
progression to overt thyroid failure, particularly if
antibodies to thyroid peroxidase are present or if the
TSH rises above 10 mU/L. In patients with non-specific
symptoms, a trial of levothyroxine therapy may be
appropriate. In those with positive autoantibodies or TSH greater than 10 mU/L, it is better to treat the thyroid failure early rather than risk loss to follow-up and subsequent presentation with profound hypothyroidism. Levothyroxine should be given in a dose sufficient to restore the serum TSH concentration to normal.
Non-thyroidal illness (‘sick euthyroidism’)
This typically presents with a low serum TSH, raised T4 and normal or low T3, in a patient with systemic illness who does not have clinical evidence of thyroid disease. These abnormalities are caused by decreased peripheralconversion of T4 to T3, altered levels of binding proteins
and their affinity for thyroid hormones, and often reduced secretion of TSH. During convalescence, serum TSH may increase to levels found in primary hypothyroidism. As thyroid function tests are difficult to interpret in patients with non-thyroidal illness, it is wise to avoid performing thyroid function tests unless there is clinical evidence of concomitant thyroid disease. If an abnormal result is found, treatment should only be given with specialist advice and the diagnosis should be re-evaluated after recovery.
Thyroid lump or swelling
A lump or swelling in the thyroid gland can be a sourceof considerable anxiety for patients. There are numerous
causes but, broadly speaking, a thyroid swelling is either
a solitary nodule, a multinodular goitre or a diffuse
goitre . Nodular thyroid disease is more common in women and occurs in approximately 30% of the adult female population. The majority of thyroid nodules are impalpable but may be identified when imaging of the neck is performed for another reason, such as during Doppler ultrasonography of the carotid arteries or computed tomographic pulmonary angiography. Increasingly, thyroid nodules are identified during staging of patients with cancer with CT, MRI or PET scans.
Palpable thyroid nodules occur in 4–8% of adult women and 1–2% of adult men, and classically present when the individual (or a friend or relative) notices a lump in the neck. Multinodular goitres and solitary nodules sometimes present with acute painful enlargement due to haemorrhage into a nodule.
Patients with thyroid nodules often worry that they have cancer, but the reality is that only 5–10% of thyroid nodules are malignant.
A solitary nodule presenting in childhood or adolescence, particularly if there is a past history of head and neck irradiation, or one presenting in the elderly should heighten suspicion of a primary thyroid malignancy . The presence of cervical
lymphadenopathy also increases the likelihood of malignancy.
Rarely, a secondary deposit from a renal, breast
or lung carcinoma presents as a painful, rapidly growing, solitary thyroid nodule.
Thyroid nodules identified on PET scanning have an approximately 33% chance of being malignant.
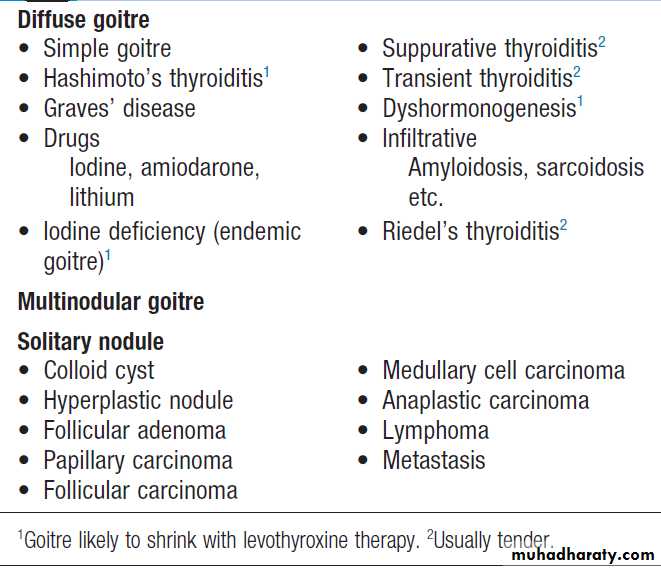
Causes of thyroid enlargement
Clinical assessment and investigationsSwellings in the anterior part of the neck most commonly
originate in the thyroid and this can be confirmed
by demonstrating that the swelling moves on swallowing.
There is a broad differential diagnosis of anterior neck
swellings, which includes lymphadenopathy, branchial
cysts, dermoid cysts and thyroglossal duct cysts (the
latter are classically located in the midline and move on
protrusion of the tongue). An ultrasound scan should be
performed urgently, if there is any doubt as to the aetiology of an anterior neck swelling.
Serum T3, T4 and TSH should be measured.
Thyroid ultrasound
If thyroid function tests are normal, an ultrasound scan
will determine the nature of the thyroid swelling. Ultrasound
can establish whether there is generalised or localised swelling of the thyroid. Inflammatory disorders
causing a diffuse goitre, such as Graves’ disease
and Hashimoto’s thyroiditis, demonstrate a diffuse
pattern of hypoechogenicity and, in the case of Graves’
disease, increased thyroid blood flow may be seen on
colour flow Doppler. The presence of thyroid autoantibodies
will support the diagnosis of Graves’ disease or
Hashimoto’s thyroiditis, while their absence in a younger
patient with a diffuse goitre and normal thyroid function
suggests a diagnosis of ‘simple goitre’ .
Ultrasound can also readily determine the size and
number of nodules within the thyroid and can distinguishsolid nodules from those with a cystic element.
It cannot reliably distinguish benign from malignant
nodules but, in experienced hands, there are some
ultrasound characteristics which are associated with a
higher likelihood of malignancy. These include: hypervascularity of the nodule, the presence of microcalcification and irregular, infiltrative margins.
A pure cystic nodule is highly unlikely to be malignant and a ‘spongiform’ appearance is also highly predicative of a benign aetiology.
Thyroid scintigraphy
Thyroid scintigraphy with 99mtechnetium should be performed in an individual with a low serum TSH and a
nodular thyroid to confirm the presence of an autonomously
functioning (‘hot’) nodule).
In such circumstances, further evaluation by fine needle
aspiration is not necessary.
‘Cold nodules’ on scintigraphy have a much higher likelihood of malignancy, but the majority are benign and so scintigraphy is not routinely used in the evaluation of thyroid nodules when TSH is normal.
Fine needle aspiration
Cytological examination of nodule, following fine needle aspiration, is recommended for most thyroid nodules >1 cm in size. Smaller nodules should be aspirated if there is a high suspicion of malignancy on clinical or ultrasound grounds, while some clinicians will be happy to observe a nodule up to 2 cm in size with a spongiform appearance. Individuals with a multinodular goitre have the same risk of malignancy as those with a solitary nodule. Sometimes, one of the nodules in a multinodular goitre is much larger than any other (a ‘dominant’ nodule), but ultimately the choice of nodule to biopsy should be based on ultrasound characteristics. Fine needle aspiration of a thyroid nodule can be performed in the outpatient clinic using 21-gauge needle and a 20 mL syringe, usually making several passes through different parts of the lesion.Ultrasound-guided needle aspiration is necessary for
impalpable nodules and to permit targeting of the solidcomponent of a mixed cystic/solid nodule. Aspiration
may be therapeutic in the small proportion of patients
in whom the swelling is a cyst, although recurrence on
more than one occasion is an indication for surgery.
Cytological examination can differentiate benign (80%)
from definitely malignant or indeterminate nodules
(20%), of which 25–50% are confirmed as cancers at
surgery.
The limitations of fine needle aspiration are that
it cannot differentiate between follicular adenoma and
carcinoma, and that in 10–20% of cases an inadequate
specimen is obtained.
Management
Solitary nodules with a solid component in which cytology either is inconclusive or shows malignant cells are
treated by surgical excision. Molecular techniques may,
in the future, improve the diagnostic accuracy of thyroid
cytology and allow a more conservative strategy for individuals with an indeterminate biopsy . Those which have benign cytology and a reassuring ultrasound appearance may by observed by interval ultrasound scans. In parts of the world with borderline low iodine intake, there is evidence that levothyroxine, in doses that suppress serum TSH, may reduce the size of some nodules. This should not be routine practice in iodine-sufficient populations.
A diffuse or multinodular goitre may also require surgical treatment for cosmetic reasons or if there is compression of local structures (resulting in stridor or dysphagia). Levothyroxine therapy may shrink the goitre of Hashimoto’s disease, particularly if serum TSH is elevated.

Molecular techniques in cytologically
indeterminate thyroid nodulesAutoimmune thyroid disease
Thyroid diseases are amongst the most prevalentantibody-mediated autoimmune diseases and are associated with other organ-specific autoimmunity. Autoantibodies may produce inflammation and destruction of thyroid tissue, resulting in hypothyroidism, goitre (in Hashimoto’s thyroiditis) or sometimes even transient thyrotoxicosis (‘Hashitoxicosis’), or they may stimulate the TSH receptor to cause thyrotoxicosis (in Graves’ disease). There is overlap between these conditions.
Graves’ disease
Most commonly affects women aged 30–50 years. The most common manifestation is thyrotoxicosis with or without a diffuse goiter, ophthalmopathy, rarely, pretibial myxoedema .
Graves’ thyrotoxicosis
PathophysiologyThe thyrotoxicosis results from the production of IgG
antibodies directed against the TSH receptor on the
thyroid follicular cell, which stimulate thyroid hormone
production and proliferation of follicular cells, leading
to goitre in the majority of patients. These antibodies are termed thyroid-stimulating immunoglobulins or TSH receptor antibodies (TRAb) and can be detected in the serum of
80–95% of patients with Graves’ disease. The concentration of TRAb in the serum is presumed to fluctuate to account for the natural history of Graves’ thyrotoxicosis .
The thyroid failure seen in some patients may result from the presence of blocking antibodies against the TSH receptor, and from tissue destruction by cytotoxic antibodies and cell-mediated immunity.
Graves’ disease has a strong genetic component.
There is 50% concordance for thyrotoxicosis between
monozygotic twins but only 5% concordance between
dizygotic twins. Genome-wide association studies have
identified polymorphisms at the MHC, CTLA4, PTPN22,
TSHR1 and FCRL3 loci as predisposing genetic variants.
Many of these loci have been implicated in the
pathogenesis of other autoimmune diseases.
A suggested trigger for the development of thyrotoxicosis
in genetically susceptible individuals may be infectionwith viruses or bacteria. Certain strains of the gut
organisms Escherichia coli and Yersinia enterocolitica
possess cell membrane TSH receptors and it has been
suggested that antibodies to these microbial antigens
may cross-react with the TSH receptors on the host
thyroid follicular cell.
In regions of iodine deficiency, iodine supplementation can precipitate thyrotoxicosis, but only in those with pre-existing subclinical Graves’ disease.
Smoking is weakly associated with Graves’ but strongly linked with the development of ophthalmopathy.
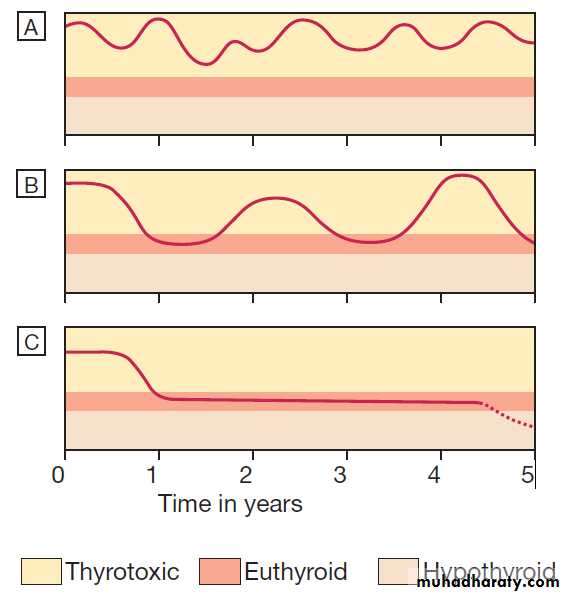
Natural history of the thyrotoxicosis of Graves’ disease.
A and B The majority (60%) of patients have either prolonged periodsof thyrotoxicosis of fluctuating severity, or periods of alternating relapse and remission.
C It is the minority who experience a single short-lived episode followed by prolonged remission and, in some cases, by the
eventual onset of hypothyroidism.
Management
Symptoms of thyrotoxicosis respond to β-blockadebut definitive treatment requires control of thyroid hormone secretion. For patients under 40 years of age, most clinicians adopt the empirical approach of prescribing a course of carbimazole and recommending surgery if relapse occurs, while 131I is employed as first or
second-line treatment in those aged over 40. A number
of observational studies have linked therapeutic 131I with
increased incidence of some malignancies, particularly
of the thyroid and gastrointestinal tract, but the results
have been inconsistent; the association may be with
Graves’ disease rather than its therapy, and the magnitude
of the effect, if any, is small.
Experience from the Chernobyl disaster suggests that younger people are more sensitive to radiation-induced thyroid cancer. In many centres, however, 131I is used extensively, even in young patients.
Antithyroid drugs.
The most commonly used are carbimazole and its active metabolite, methimazole (not available in the UK). Propylthiouracil is equally effective.
These drugs reduce the synthesis of new thyroid hormones by inhibiting the iodination of tyrosine.
Carbimazole also has an immunosuppressive action, leading to a reduction in serum TRAb concentrations, but this is not enough to influence the natural history of the thyrotoxicosis significantly.
Antithyroid drugs should be introduced at high
doses (carbimazole 40–60 mg daily or propylthiouracil
400–600 mg daily). Usually, this results in subjective
improvement within 10–14 days and renders the patient
clinically and biochemically euthyroid at 3–4 weeks.
At this point, the dose can be reduced and titrated to maintain T4 and TSH within their reference range.
In most patients, carbimazole is continued at 5–20 mg per day for 12–18 months in the hope that remission will occur.
Patients with thyrotoxicosis relapse in at least 50% of
cases, usually within 2 years of stopping treatment.
Rarely, T4 and TSH levels fluctuate between those of
thyrotoxicosis and hypothyroidism at successive review
appointments, despite good drug compliance, presumably
due to rapidly changing concentrations of TRAb. In
these patients, satisfactory control can be achieved by
blocking thyroid hormone synthesis with carbimazole
30–40 mg daily and adding levothyroxine 100–150 μg daily as replacement therapy (a ‘block and replace’ regime).
Antithyroid drugs can have adverse effects. The most
common is a rash. Agranulocytosis is a rare but potentiallyserious complication that cannot be predicted by routine measurement of white blood cell count, but which is reversible on stopping treatment. Patients should be warned to stop the drug and seek medical advice immediately, should a severe sore throat or fever develop whilst on treatment. Propylthiouracil is associated with a small but definite risk of hepatotoxicity, which, in some instances, has resulted in liver failure requiring liver transplantation, and even in death. It should, therefore, be considered second-line therapy to carbimazole and only be used during pregnancy or breastfeeding (see below), or if an adverse reaction to carbimazole has occurred.
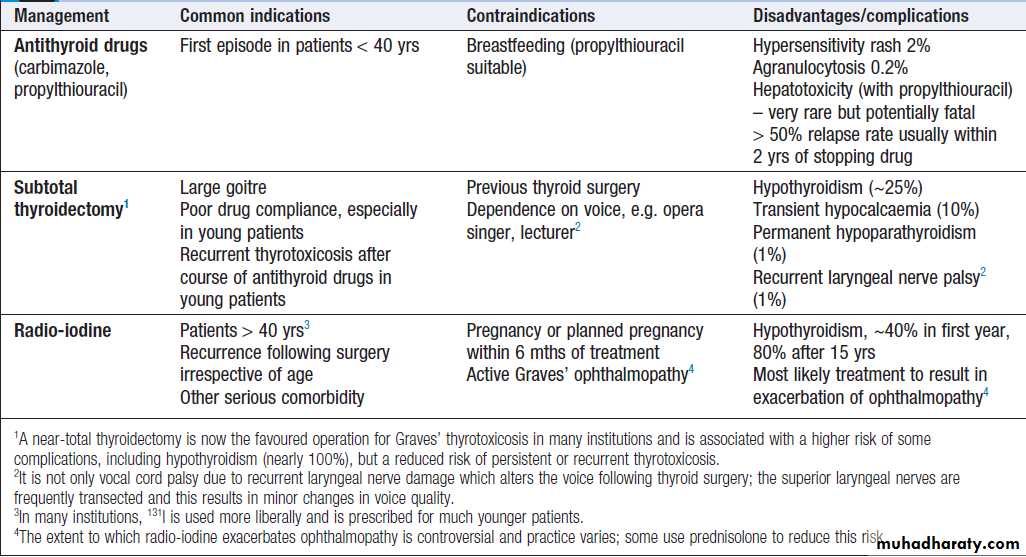
Comparison of treatments for the thyrotoxicosis of Graves’ disease
Thyroid surgery. Patients should be rendered euthyroidwith antithyroid drugs before operation. Potassium
iodide, 60 mg 3 times daily orally, is often added for
2 weeks before surgery to inhibit thyroid hormone
release and reduce the size and vascularity of the gland,
making surgery technically easier. Traditionally, a ‘subtotal’
thyroidectomy is performed, in which a portion
of one lobe of the thyroid is left in situ, with the aim
of rendering the patient euthyroid post-operatively.
While complications of surgery are rare and 80% of
patients are euthyroid, 15% are permanently hypothyroid
and 5% remain thyrotoxic.
As a consequence, many endocrine surgeons now opt to perform a ‘neartotal’ thyroidectomy, leaving behind only a small portion of gland adjacent to the recurrent laryngeal nerves.
This strategy invariably results in permanent hypothyroidism and is probably associated with a higher risk of hypoparathyroidism, but maximises the potential
for cure of thyrotoxicosis.
Radioactive iodine. 131I is administered orally as a single
dose, and is trapped and organified in the thyroid . Although 131I decays within a few weeks, it has long-lasting inhibitory effects on survival and replication of follicular cells. The variable radioiodine uptake and radiosensitivity of the gland means that the choice of dose is empirical; in most centres, approximately 400 MBq (10 mCi) is given orally. This regimen is effective in 75% of patients within
4–12 weeks. During the lag period, symptoms can be
controlled by a β-blocker or, in more severe cases, by
carbimazole. However, carbimazole reduces the efficacy
of 131I therapy because it prevents organification of 131I in
the gland, and so should be avoided until 48 hours after
radio-iodine administration.
If thyrotoxicosis persists after 6 months, a further dose of 131I can be given. The disadvantage of 131I treatment is that the majority of patients eventually develop hypothyroidism. 131I is usually avoided in patients with Graves’ ophthalmopathy and evidence of significant active orbital inflammation.
It can be administered with caution in those with mild or ‘burnt-out’ eye disease, when it is customary to cover the treatment with a 6-week tapering course of oral prednisolone. In women of reproductive age, pregnancy must be excluded before administration of 131I and avoided for 6 months thereafter; men are also advised against fathering children for 6 months after receiving 131I.
Thyrotoxicosis in pregnancy
The coexistence of pregnancy and thyrotoxicosis is unusual, as anovulatory cycles are common in thyrotoxic patients and autoimmune disease tends to remit during pregnancy, when the maternal immune response is suppressed. Thyroid function tests must be interpreted in the knowledge that TBG, and hence total T4 and T3 levels, are increased in pregnancy and that TSH reference ranges may be lower; a fully suppressed TSH with elevated free thyroid hormone levels indicates thyrotoxicosis. The thyrotoxicosis is almost always caused by Graves’ .Both mother and fetus must be considered, since maternal hormones, TRAb and antithyroid drugs can all cross the placenta to some degree, exposing the fetus to the risks of thyrotoxicosis, iatrogenic hypothyroidism and goitre.Poorly controlled maternal thyrotoxicosis can result in fetal tachycardia, intrauterine growth retardation, prematurity, stillbirth and possibly even congenital malformations.
Antithyroid drugs are the treatment of choice for
thyrotoxicosis in pregnancy. Carbimazole has been
associated with rare cases of embryopathy, particularly
a skin defect known as aplasia cutis, and should be avoided in the first trimester. Propylthiouracil should be used in its place but, because of its potential hepatotoxicity, should be replaced with carbimazole from the beginning of the second trimester. Both drugs cross the placenta and will effectively treat thyrotoxicosis in the fetus caused by transplacental passage of TRAb.
To avoid fetal hypothyroidism (which could affect brain development) and a resultant goitre, it is important to use the smallest dose of antithyroid drug (optimally, less
than 150 mg propylthiouracil or 15 mg carbimazole per
day) that will maintain maternal (and presumably fetal)
free T4, T3 and TSH within their respective reference
ranges. Frequent review of mother and fetus (monitoring
heart rate and growth) is important. TRAb levels can
be measured in the third trimester to predict the likelihood
of neonatal thyrotoxicosis. When TRAb levels are
not elevated, the antithyroid drug can be discontinued
4 weeks before the expected date of delivery to minimise
the risk of fetal hypothyroidism at the time of maximum
brain development.
After delivery, if antithyroid drug is required and the patient wishes to breastfeed, then propylthiouracil is the drug of choice, as it is excreted in the milk to a much lesser extent than carbimazole. Thyroid function should be monitored periodically in the breastfed child.
If thyroid surgery is necessary because of poor drug
compliance or drug hypersensitivity, it is most safely
performed in the second trimester.
Radioactive iodine is absolutely contraindicated, as it invariably induces fetal hypothyroidism.

Thyrotoxicosis in adolescence
Graves’ ophthalmopathy
This condition is immunologically mediated but theautoantigen has not been identified. Within the orbit
(and the dermis) there is cytokine-mediated proliferation
of fibroblasts which secrete hydrophilic glycos-aminoglycans.
The resulting increase in interstitial fluid content, combined with a chronic inflammatory cell infiltrate, causes marked swelling and ultimately fibrosis of the extraocular muscles and a rise in retrobulbar pressure.
The eye is displaced forwards (proptosis, exophthalmos) and in severe cases there is optic nerve compression.
Ophthalmopathy, like thyrotoxicosis , typically follows an episodic course and it is helpful to distinguish patients with active inflammation (periorbital oedema and conjunctival inflammation with changing orbital signs) from those in whom the inflammation has ‘burnt out’. Eye disease is detectable in up to 50% of thyrotoxic patients at presentation, but active ocular inflammation may occur before or after thyrotoxic episodes (exophthalmic Graves’ disease). It is more common in cigarette smokers and is exacerbated by poor control of thyroid function, especially hypothyroidism. The most frequent presenting symptoms are related to increased exposure of the cornea, resulting from proptosis and lid retraction.
There may be excessive lacrimation made worse by wind and bright light, a ‘gritty’ sensation in the eye, and pain due to conjunctivitis or corneal ulceration.
In addition, there may be reduction of visual acuity and/or visual fields as a consequence of corneal oedema or optic nerve compression.
Other signs of optic nerve compression include reduced colour vision and a relative afferent pupillary defect .
If the extraocular muscles are involved and do not act in concert, diplopia results.
The majority of patients require no treatment other
than reassurance. Smoking cessation should be activelyencouraged. Methylcellulose eye drops and gel counter
the gritty discomfort of dry eyes, and tinted glasses or
side shields attached to spectacle frames reduce the
excessive lacrimation triggered by sun or wind. In
patients with mild Graves’ ophthalmopathy,
oral selenium (100 μg twice daily for 6 months) improves quality of life, reduces ocular involvement and slows progression of disease; the mechanism of action is not known but may relate to an antioxidant effect.
More severe inflammatory episodes are treated
with glucocorticoids (e.g. daily oral prednisolone or
pulsed IV methylprednisolone) and sometimes orbital
radiotherapy. There is also an increasing trend to use
immunosuppressant therapies, such as ciclosporin, in
combination with glucocorticoids.
Loss of visual acuity is an indication for urgent surgical decompression of the orbit. In ‘burnt-out’ disease, surgery to the eyelids and/or ocular muscles may improve conjunctival exposure, cosmetic appearance and diplopia
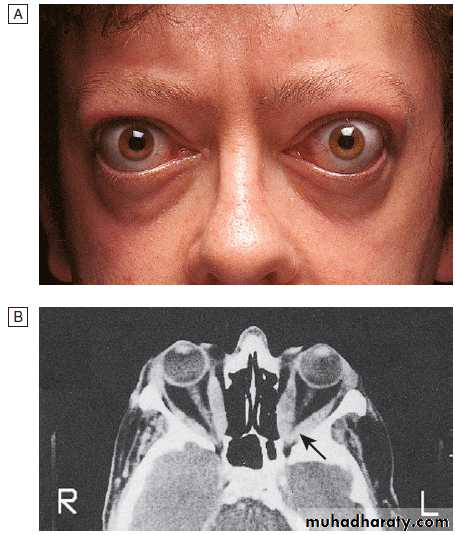
Graves’ disease.
A Bilateral ophthalmopathy in a 42-year-old man. The main symptoms were diplopia in all directions of gaze and reduced visual acuity in the left eye. The periorbital swelling is due to retrobulbar fat prolapsing into the eyelids, and increased interstitial fluid as a result of raised intraorbital pressure.B Transverse CT of the
orbits, showing the enlarged extraocular muscles. This is most obvious at the apex of the left orbit (arrow), where compression of the optic nerve caused reduced visual acuity.

Effects of selenium supplementation in
mild Graves’ ophthalmopathyPretibial myxoedema
This infiltrative dermopathy occurs in fewer than 10%
of patients with Graves’ disease and has similar pathological features as occur in the orbit. It takes the form of raised pink- coloured or purplish plaques on the anterior aspect of the leg, extending on to the dorsum of the foot.
The lesions may be itchy and the skin may have
a ‘peau d’orange’ appearance with growth of coarse
hair; less commonly, the face and arms are affected.
Treatment is rarely required, but in severe cases topical
glucocorticoids may be helpful.
Hashimoto’s thyroiditis
Hashimoto’s thyroiditis is characterised by destructivelymphoid infiltration of the thyroid, ultimately leading
to a varying degree of fibrosis and thyroid enlargement.
There is an increased risk of thyroid lymphoma , although this is exceedingly rare. The nomenclature of autoimmune hypothyroidism is confusing. Some authorities reserve the term ‘Hashimoto’s thyroiditis’ for patients with positive antithyroid peroxidase autoantibodies and a firm goitre who may or may not be hypothyroid, and use the term ‘spontaneous atrophic hypothyroidism’ for hypothyroid patients without a goitre in whom TSH receptor-blocking antibodies may be more important than antiperoxidase antibodies. However, these syndromes can both be considered as variants of the same underlying disease process.
Hashimoto’s thyroiditis increases in incidence with
age and affects approximately 3.5 per 1000 women and0.8 per 1000 men each year. Many present with a small
or moderately sized diffuse goitre, which is characteristically firm or rubbery in consistency. The goitre may be soft, however, and impossible to differentiate from
simple goitre by palpation alone.
Around 25% of patients are hypothyroid at presentation. In the remainder, serum T4 is normal and TSH normal or raised, but these patients are at risk of developing overt hypothyroidism in future years.
Antithyroid peroxidase antibodies are present in the serum in more than 90% of patients with Hashimoto’s thyroiditis. In those under the age of 20 years, antinuclear factor (ANF) may also be positive.
Levothyroxine therapy is indicated as treatment for
hypothyroidism and also to shrink an associated goitre. In this context, the dose of thyroxine should be sufficient to suppress serum TSH to low but detectable levels.
Transient thyroiditis
Subacute (de Quervain’s) thyroiditisIn its classical painful form, subacute thyroiditis is a
transient inflammation of the thyroid gland occurring
after infection with Coxsackie, mumps or adenoviruses.
There is pain in the region of the thyroid that may radiate to the angle of the jaw and the ears, and is made worse by swallowing, coughing and movement of the neck. The thyroid is usually palpably enlarged and tender. Systemic upset is common. Affected patients are usually females aged 20–40 years. Painless transient thyroiditis can also occur after viral infection and in patients with underlying autoimmune disease. The condition can also be precipitated by drugs, including interferon-α and lithium.
Irrespective of the clinical presentation, inflammation
in the thyroid gland occurs and is associated with releaseof colloid and stored thyroid hormones, but also with damage to follicular cells and impaired synthesis of new thyroid hormones. As a result, T4 and T3 levels are raised for 4–6 weeks until the pre-formed colloid is depleted. Thereafter, there is usually a period of hypothyroidism of variable severity before the follicular cells recover and normal thyroid function is restored within 4–6 months . In the thyrotoxic phase, the iodine uptake is low, because the damaged follicular cells are unable to trap iodine and because TSH secretion is suppressed. Low-titre thyroid autoantibodies appear transiently in the serum, and the erythrocyte sedimentation rate (ESR) is usually raised.
High-titre autoantibodies suggest an underlying autoimmune pathology and greater risk of recurrence and ultimate progression to hypothyroidism.
The pain and systemic upset usually respond to
simple measures such as non-steroidal anti-inflammatory
drugs (NSAIDs). Occasionally, however, it may be
necessary to prescribe prednisolone 40 mg daily for
3–4 weeks. The thyrotoxicosis is mild and treatment
with a β-blocker is usually adequate. Antithyroid drugs
are of no benefit because thyroid hormone synthesis
is impaired rather than enhanced.
Careful monitoring of thyroid function and symptoms is required so that levothyroxine can be prescribed temporarily in the hypothyroid phase.
Care must be taken to identify patients presenting with hypothyroidism who are in the later stages of a transient thyroiditis, since they are unlikely to require life-long levothyroxine therapy .
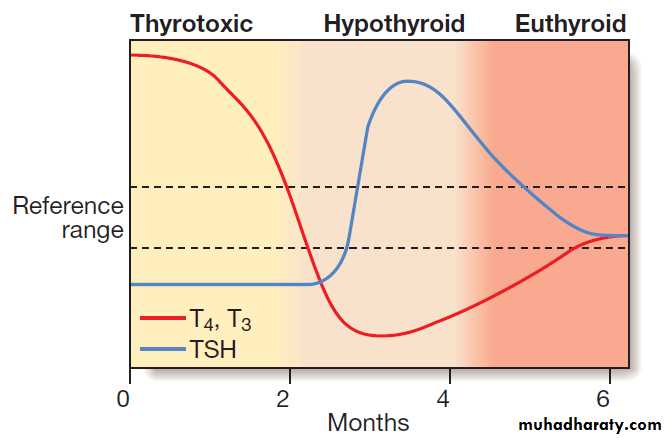
Thyroid function tests in an episode of transient
thyroiditis. This pattern might be observed in classical subacute(de Quervain’s) thyroiditis, painless thyroiditis or post-partum thyroiditis.
The duration of each phase varies between patients.
Post-partum thyroiditis
The maternal immune response, which is modified during pregnancy to allow survival of the fetus, is enhanced after delivery and may unmask previously unrecognised subclinical autoimmune thyroid disease. Surveys have shown that transient biochemical disturbances of thyroid function occur in 5–10% of women within 6 months of delivery . Those affected are likely to have anti-thyroid peroxidase antibodies in the serum in early pregnancy.Symptoms of thyroid dysfunction are rare and there is no association between postnatal depression and abnormal thyroid function tests.
However, symptomatic thyrotoxicosis presenting for the first time within 12 months of childbirth is likely to be due to post-partum thyroiditis and the diagnosis is confirmed by a negligible radio-isotope uptake. The clinical course and treatment are similar to those of painless subacute thyroiditis .
Postpartum thyroiditis tends to recur after subsequent pregnancies, and eventually patients progress over a period of years to permanent hypothyroidism.
Iodine-associated thyroid disease
Iodine deficiencyThyroid enlargement is extremely common in certain
mountainous parts of the world, such as the Andes, the
Himalayas and central Africa, where there is dietary
iodine deficiency (endemic goitre). Most patients are
euthyroid with normal or raised TSH levels, although
hypothyroidism can occur with severe iodine deficiency.
Iodine supplementation programmes have abolished
this condition in most developed countries.
Iodine-induced thyroid dysfunction
Iodine has complex effects on thyroid function. Very
high concentrations of iodine inhibit thyroid hormone
release and this forms the rationale for iodine treatment
of thyroid storm and prior to thyroid surgery for
thyrotoxicosis . Iodine administration initially
enhances, but then inhibits, iodination of tyrosine and
thyroid hormone synthesis .
The resulting effect of iodine on thyroid function varies
according to whether the patient has an iodine-deficient
diet or underlying thyroid disease. In iodine-deficient
parts of the world, transient thyrotoxicosis may be precipitated by prophylactic iodinisation programmes.
In iodine-sufficient areas, thyrotoxicosis can be precipitated
by radiographic contrast medium or expectorantsin individuals who have underlying thyroid disease predisposing to thyrotoxicosis, such as multinodular goitre
or Graves’ disease in remission.
Induction of thyrotoxicosis by iodine is called the
Jod – Basedow effect.
Chronic excess iodine administration can, however, result in hypothyroidism.
Increased iodine within the thyroid gland down-regulates iodine trapping, so that uptake is low in all circumstances.
Amiodarone
The anti-arrhythmic agent amiodarone has a structure
that is analogous to that of T4 and contains huge amounts of iodine; a 200 mg dose contains 75 mg iodine, compared with a daily dietary requirement of just 125 μg. Amiodarone also has a cytotoxic effect on thyroid follicular cells and inhibits conversion of T4 to T3. Most patients receiving amiodarone have normal thyroid function, but up to 20% develop hypothyroidism or thyrotoxicosis and so thyroid function should be monitored regularly.
The ratio of T4:T3 is elevated and TSH provides the best indicator of thyroid function.
The thyrotoxicosis can be classified as either:
• type I: iodine-induced excess thyroid hormonesynthesis in patients with an underlying thyroid
disorder, such as nodular goitre or latent Graves’
disease
• type II: thyroiditis due to a direct cytotoxic effect if
amiodarone administration results in a transient
thyrotoxicosis.
These patterns can overlap and can be difficult to
distinguish clinically, as iodine uptake is low in both.
There is no widely accepted management algorithm,
although the iodine excess renders the gland resistant to
radio-iodine.
Antithyroid drugs may be effective in patients with the
type I form, but are ineffective in type II thyrotoxicosis. Prednisolone is beneficial in the type II form. A pragmatic approach is to commence combination therapy with an antithyroid drug and glucocorticoid in patients with significant thyrotoxicosis. A rapid response (within 1–2 weeks) usually indicates a type II picture and permits withdrawal of the antithyroid therapy; a slower response suggests a type I picture, when antithyroid drugs may be continued and prednisolone withdrawn. Potassium perchlorate can also be used to inhibit iodine trapping in the thyroid. If the cardiac state allows, amiodarone should be discontinued, but it has a long half-life (50–60 days) and so its effects are long-lasting.To minimise the risk of type I thyrotoxicosis, thyroid function should be measured in all patients prior to commencement of amiodarone therapy, and amiodarone should be avoided if TSH is suppressed.
Hypothyroidism should be treated with levothyroxine,
which can be given while amiodarone is continued.
Simple diffuse goitre
This form of goitre usually presents between the ages of15 and 25 years, often during pregnancy, and tends to
be noticed by friends and relatives rather than the patient. Occasionally, there is a tight sensation in the neck, particularly when swallowing. The goitre is soft and symmetrical, and the thyroid enlarged to two or three times normal. There is no tenderness, lymphadenopathy or overlying bruit. Concentrations of T3, T4 and TSH are normal and no thyroid autoantibodies are detected in the serum. No treatment is necessary and the goitre usually regresses. In some, however, the unknown stimulus to thyroid enlargement persists and, as a result of recurrent episodes of hyperplasia and involution during the following 10–20 years, the gland becomes multinodular with areas of autonomous function.
Multinodular goitre
Patients with thyroid enlargement in the absence of thyroid dysfunction or positive autoantibodies (i.e. with ‘simplegoitre’, see above) as young adults may progress to
develop nodules. These nodules grow at varying rates
and secrete thyroid hormone ‘autonomously’, thereby
suppressing TSH-dependent growth and function in the
rest of the gland. Ultimately, complete suppression of
TSH occurs in about 25% of cases, with T4 and T3 levels
within the reference range(subclinical thyrotoxicosis )
but sometimes elevated (toxic multinodular goitre).
Clinical features and investigations
Multinodular goitre is usually diagnosed in patientspresenting with thyrotoxicosis, a large goitre with or
without tracheal compression, or sudden painful swelling
caused by haemorrhage into a nodule or cyst. The
goitre is nodular or lobulated on palpation and may
extend retrosternally; however, not all multinodular
goitres causing thyrotoxicosis are easily palpable. Very
large goitres may cause mediastinal compression with
stridor , dysphagia and obstruction of the superior vena cava. Hoarseness due to recurrent laryngeal nerve palsy can occur, but is far more suggestive of thyroid carcinoma.
The diagnosis can be confirmed by ultrasonography
and/or thyroid scintigraphy . In patients with large goitres, a flow-volume loop is a good screening test for significant tracheal compression .
If intervention is contemplated, a CT or MRI of the thoracic inlet should be performed to quantify the degree of tracheal displacement or compression and the extent of retrosternal extension.
Nodules should be evaluated for the possibility of thyroid neoplasia.
Management
If the goitre is small, no treatment is necessary butannual thyroid function testing should be arranged, as
the natural history is progression to a toxic multinodular
goitre. Thyroid surgery is indicated for large goitres
which cause mediastinal compression or which are
cosmetically unattractive. 131I can result in a significant
reduction in thyroid size and may be of value in elderly patients. Levothyroxine therapy is of no benefit in shrinking multinodular goitres in iodine-sufficient countries and may simply aggravate any associated thyrotoxicosis.
In toxic multinodular goitre, treatment is usually
with 131I. The iodine uptake is lower than in Graves’disease, so a higher dose may be administered (up
to 800 Mbq (approximately 20 mCi)) and hypothyroidism
is less common. In thyrotoxic patients with a large goitre, thyroid surgery may be indicated.
Long-term treatment with antithyroid drugs is not
usually employed, as relapse is invariable after drug
withdrawal.
Asymptomatic patients with subclinical thyrotoxicosis
are increasingly being treated with 131I on the grounds that a suppressed TSH is a risk factor for atrial fibrillation and, particularly in post-menopausal women, osteoporosis.
Thyroid neoplasia
Patients with thyroid tumours usually present with a
solitary nodule . Most are benign and a few of
these, called ‘toxic adenomas’, secrete excess thyroid
hormones. Primary thyroid malignancy is rare, accounting
for less than 1% of all carcinomas, and has an incidence
of 25 per million per annum. As shown in Box ,
it can be classified according to the cell type of
origin. With the exception of medullary carcinoma,
thyroid cancer is more common in females.
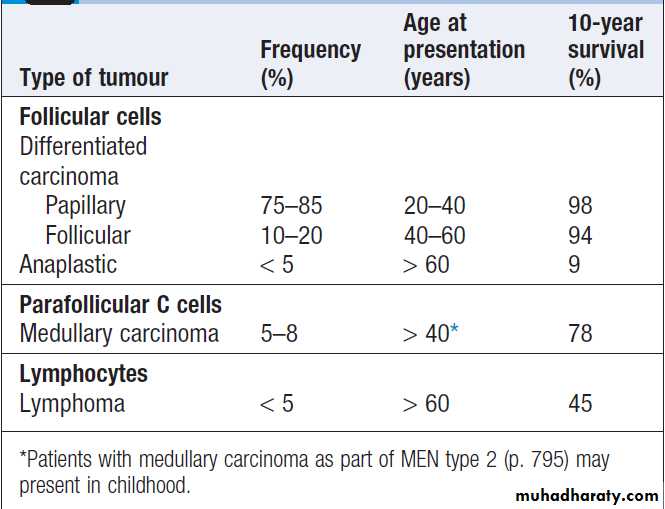
Malignant thyroid tumours
Toxic adenomaA solitary toxic nodule is the cause of less than 5% of
all cases of thyrotoxicosis. The nodule is a follicular
adenoma, which autonomously secretes excess thyroid
hormones and inhibits endogenous TSH secretion, with
subsequent atrophy of the rest of the thyroid gland. The
adenoma is usually greater than 3 cm in diameter.
Most patients are female and over 40 years of age.
Although many nodules are palpable, the diagnosis can
be made with certainty only by thyroid scintigraphy .
The thyrotoxicosis is usually mild and in almost 50% of patients the plasma T3 alone is elevated (T3 thyrotoxicosis). 131I (400–800 MBq (10–20 mCi)) is highly effective and is an ideal treatment since the atrophic cells surrounding the nodule do not take up iodine and so receive little or no radiation. For this reason, permanent hypothyroidism is unusual. A surgical hemithyroidectomy is an alternative.
Differentiated carcinoma
Papillary carcinomaThis is the most common of the malignant thyroid
tumours and accounts for 90% of irradiation-induced
thyroid cancer. It may be multifocal and spread is initially
to regional lymph nodes. Some patients present
with cervical lymphadenopathy and no apparent thyroid
enlargement; in such instances, the primary lesion may
be less than 10 mm in diameter.
Follicular carcinoma
This is always a single encapsulated lesion. Spread to
cervical lymph nodes is rare. Metastases are blood-borne
and are most often found in bone, lungs and brain.
Management
This is usually by total thyroidectomy followed by a large dose of 131I (3700 MBq (approximately 100 mCi)) in order to ablate any remaining thyroid tissue, normal or malignant. Recent data indicate that a 131I dose of 1100 MBq (approximately 30 mCi) may be equally as effective at thyroid ablation . Thereafter, long-term treatment with levothyroxine in a dose sufficient to suppress TSH (usually 150–200 μg daily) is important, as there is evidence that growth of differentiated thyroid carcinomas is TSH-dependent. Follow-up is by measurement of serum thyroglobulin, which should be undetectable in patients whose normal thyroid has been ablated and who are taking a suppressive dose of levothyroxine.Detectable thyroglobulin is suggestive of tumour recurrence or metastases, which may be localised by US, CT, MRI or whole-body scanning with 131I, and may respond to further radio-iodine therapy. Radio-iodine treatment in
thyroid cancer and isotope scanning both require serum
TSH concentrations to be elevated (> 20 mU/L). This
may be achieved by stopping levothyroxine for 4–6 weeks, inducing symptomatic hypothyroidism, or by administering intramuscular injections of recombinant human TSH. Patients usually find the latter approach preferable but it is more expensive. Clinical trials are currently ongoing with novel anti-cancer agents, such as tyrosine kinase inhibitors, in patients with advanced papillary and follicular carcinoma that is refractive to radio-iodine.

Ablative radio-iodine following thyroidectomy for differentiated thyroid cancer
PrognosisMost patients with papillary and thyroid cancer will be
cured with appropriate treatment. Adverse prognostic
factors include older age at presentation, the presence
of distant metastases, male sex and the identification
of certain histological subtypes. However, radio-iodine
therapy can be effective in treating even those with
distant metastases, particularly small-volume disease in
the lungs, and so prolonged survival is quite common.
Anaplastic carcinoma and lymphoma
These two conditions are difficult to distinguish clinicallybut are distinct cytologically and histologically.Patients are usually over 60 years of age and present with rapid thyroid enlargement over 2–3 months. The goitre is hard and there may be stridor due to tracheal compression and hoarseness due to recurrent laryngeal nerve palsy. There is no effective treatment for anaplastic carcinoma, although surgery and radiotherapy may be considered in some. In older patients, median survival is only 7 months.
The prognosis for lymphoma, which may arise from pre-existing Hashimoto’s thyroiditis, is better , with a median survival of 9 years. Some 98% are non- Hodgkin’s lymphomas, usually the diffuse large B-cell subtype.
Treatment is with combination chemotherapy, such as the CHOP regime and external beam radiotherapy.
Medullary carcinoma
This tumour arises from the parafollicular C cells of the
thyroid. In addition to calcitonin, the tumour may secrete 5-hydroxytryptamine (5-HT, serotonin), various peptides of the tachykinin family, adrenocorticotrophic hormone (ACTH) and prostaglandins. As a consequence, carcinoid and Cushing’s syndrome may occur. Patients usually present in middle age with a firm thyroid mass. Cervical lymph node involvement is common but distant metastases are rare initially. Serum calcitonin levels are raised and are useful in monitoring response to treatment. Despite the very high levels of calcitonin found in some patients, hypocalcaemia is extremely rare; however, hypercalcitoninaemia can be associated with severe, watery diarrhoea.
Treatment is by total thyroidectomy with removal of regional cervical lymph nodes. Since the C cells do not concentrate iodine and are not responsive to TSH, there is no role for 131I therapy or TSH suppression with levothyroxine.
External beam radiotherapy may be considered in some patients at high risk of local recurrence.
Vandetanib, a tyrosine kinase inhibitor, is licensed for patients with advanced medullary cancer.
The prognosis is less good than for papillary and follicular carcinoma, but individuals can live for many decades with persistent disease which behaves in an indolent fashion. Medullary carcinoma of the thyroid may occur sporadically, or in families as part of the MEN type 2 syndrome .
Riedel’s thyroiditis
This is not a form of thyroid cancer, but the presentationis similar and the differentiation can usually only be
made by thyroid biopsy. It is an exceptionally rare
condition of unknown aetiology, in which there is
extensive infiltration of the thyroid and surrounding
structures with fibrous tissue. There may be associated
mediastinal and retroperitoneal fibrosis. Presentation
is with a slow-growing goitre which is irregular and
stony-hard.
There is usually tracheal and oesophageal compression necessitating partial thyroidectomy. Other recognised complications include recurrent laryngeal nerve palsy, hypoparathyroidism and eventually hypothyroidism.
Congenital thyroid disease
Early treatment with levothyroxine is essential to prevent irreversible brain damage in children (cretinism) with congenital hypothyroidism. Routine screening of TSH levels in heelprick blood samples obtained 5–7 days after birth (as part of the Guthrie test) has revealed an incidence of approximately 1 in 3000, resulting from thyroid agenesis, ectopic or hypoplastic glands, or dyshormonogenesis. Congenital hypothyroidism is thus six times more common than phenylketonuria. It is now possible to start thyroid replacement therapy within 2 weeks of birth. Developmental assessment of infants treated at this early stage has revealed no differences between cases and controls in most children.
Dyshormonogenesis
Several autosomal recessive defects in thyroid hormonesynthesis have been described; the most common
results from deficiency of the intrathyroidal peroxidase enzyme. Homozygous individuals present with congenital
hypothyroidism; heterozygotes present in the first two decades of life with goitre, normal thyroid hormone levels and a raised TSH.
The combination of dyshormonogenetic goitre and nerve deafness is known as Pendred’s syndrome and is due to mutations in pendrin, the protein which transports iodide to the luminal surface of the follicular cell .
Thyroid hormone resistance
This is a rare disorder in which the pituitary andhypothalamus are resistant to feedback suppression of
TSH by T3, sometimes due to mutations in the thyroid
hormone receptor β or because of defects in monodeiodinase activity.
The result is high levels of TSH, T4 and T3, often with a moderate goitre which may not be noted until adulthood. Thyroid hormone signalling is highly complex and involves different isozymes of both monodeiodinases and thyroid hormone receptors in different tissues.
For that reason, other tissues may or may not share the resistance to thyroid hormone and there may be features of thyrotoxicosis (e.g. tachycardia).
This condition can be difficult to distinguish from
an equally rare TSH-producing pituitary tumour; administration of TRH results in elevation of TSH in thyroid hormone resistance and not in TSHoma,
but an MRI scan of the pituitary may be necessary to exclude a macroadenoma.
The thyroid gland in old age
Thyrotoxicosis
• Causes: commonly due to multinodular goitre.
• Clinical features: apathy, anorexia, proximal myopathy, atrial fibrillation and cardiac failure predominate.
• Non-thyroidal illness: thyroid function tests interpretation may be altered by intercurrent illness.
Hypothyroidism
• Clinical features:non-specific, such as physical and mental slowing, are often attributed to increasing age and the diagnosis is delayed.
• Myxoedema coma : more likely in the elderly.
• Levothyroxine dose: to avoid exacerbating latent or established heart disease, the starting dose should be 25 μg daily. Levothyroxine requirements fall with increasing age and few patients need more than 100 μg daily.
• Other medication : may interfere with absorption or metabolism of levothyroxine, necessitating an increase in dose.

Thyroid disease in pregnancy

Thyroid disease in pregnancy
THE REPRODUCTIVE SYSTEM
Clinical practice in reproductive medicine is shared
between several specialties, including gynaecology,
urology, paediatrics, psychiatry and endocrinology. The
following section is focused on disorders managed by
endocrinologists.
Functional anatomy, physiology and investigations
The physiology of male and female reproductive function
is illustrated in Figures.
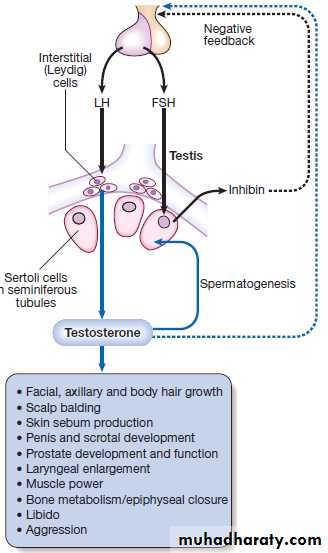
Male reproductive physiology.
(FSH = follicle-stimulatinghormone; LH = luteinising hormone).
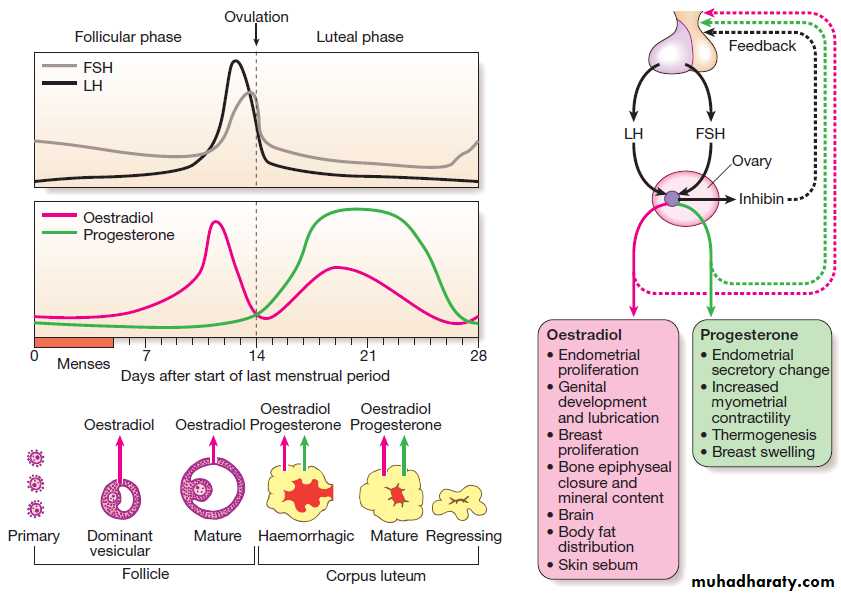
Female reproductive physiology and the normal menstrual cycle.
The male
In the male, the testis subserves two principal functions:
synthesis of testosterone by the interstitial Leydig cells
under the control of luteinising hormone (LH), and
spermatogenesis by Sertoli cells under the control of
follicle-stimulating hormone (FSH) (but also requiring
adequate testosterone).
Negative feedback suppression of LH is mediated principally by testosterone, while secretion of another hormone by the testis, inhibin, suppresses FSH. The axis can be assessed easily by a random blood sample for testosterone, LH and FSH. Testosterone levels are higher in the morning and therefore, if testosterone is marginally low, sampling should be repeated in the early morning (0900 hrs).
Testosterone is largely bound in plasma to sex hormone-binding globulin, and this can also be measured to calculate the ‘free androgen index’ or the ‘bioavailable’ testosterone.
Testicular function can also be tested by semen analysis.
There is no equivalent of the menopause in men,
although testosterone concentrations decline slowly
from the fourth decade onwards.
The female
In the female, physiology varies during the normal menstrual cycle. FSH stimulates growth and development of ovarian follicles during the first 14 days after the menses. This leads to a gradual increase in oestradiol production from granulosa cells, which initially suppresses FSH secretion (negative feedback) but then, above a certain level, stimulates an increase in both the frequency and amplitude of gonadotrophin-releasing hormone (GnRH) pulses, resulting in a marked increase in LH secretion (positive feedback).The mid-cycle ‘surge’ of LH induces ovulation. After release of the ovum, the follicle differentiates into a corpus luteum, which secretes progesterone.
Unless pregnancy occurs during the cycle, the corpus luteum regresses and the fall in progesterone levels results in menstrual bleeding. Circulating levels of oestrogen and progesterone in pre-menopausal women are, therefore, critically dependent on the time of the cycle. The most useful ‘test’ of ovarian function is a careful menstrual history:
if menses are regular, measurement of gonadotrophins and oestrogen is not necessary. In addition, ovulation can be confirmed by measuring plasma progesterone levels during the luteal phase (‘day 21 progesterone’).
Cessation of menstruation (the menopause) occurs at an average age of approximately 50 years in developed countries. In the 5 years before, there is a gradual increase in the number of anovulatory cycles and this is referred to as the climacteric.
Oestrogen and inhibin secretion falls and negative feedback results in increased pituitary secretion of LH and FSH (typically to levels >30 U/L (3.3 μg/L)).
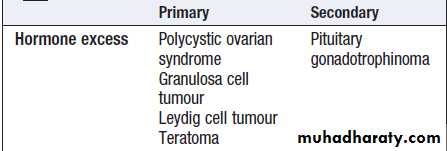
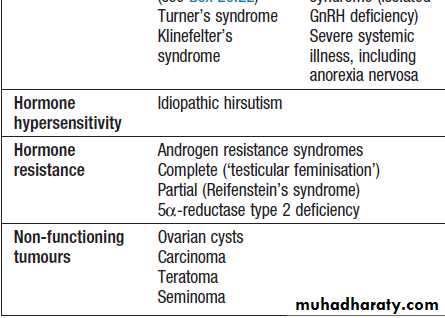


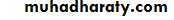
Classification of diseases of
the reproductive systemPresenting problems in reproductive disease
Delayed pubertyPuberty is considered to be delayed if the onset of the
physical features of sexual maturation has not occurred
by a chronological age that is 2.5 standard deviations
(SD) above the national average. In the UK, this is by the
age of 14 in boys and 13 in girls. Genetic factors have a
major influence in determining the timing of the onset
of puberty, such that the age of menarche (the onset of
menstruation) is often comparable within sibling and
mother–daughter pairs and within ethnic groups.
However, because there is also a threshold for body
weight that acts as a trigger for normal puberty, the
onset of puberty can be influenced by other factors
including nutritional status and chronic illness .
Clinical assessment
The key issue is to determine whether the delay in puberty
is simply because the ‘clock is running slow’ (constitutional delay of puberty) or because there is pathology in the hypothalamus/pituitary (hypogonadotrophic hypogonadism) or the gonads (hypergonadotrophic hypogonadism). A general history and physical examination should be performed with particular reference to previous or current medical disorders, social circumstances and family history. Body proportions, sense
of smell and pubertal stage should be carefully documented and, in boys, the presence or absence of testes
in the scrotum noted. Current weight and height may be
plotted on centile charts, along with parental heights.
Previous growth measurements in childhood, which can
usually be obtained from health records, are extremelyuseful. Healthy growth usually follows a centile. Usually,
children with constitutional delay have always been
small, but have maintained a normal growth velocity
that is appropriate for bone age. Poor linear growth,
with ‘crossing of the centiles’, is more likely to be associated
with acquired disease. Issues that are commonly
encountered in the management of adolescents with
delayed puberty are summarised in Box.
Causes of delayed puberty and Hypogonadism
Constitutional delayHypogonadotrophic hypogonadism
• Structural hypothalamic/pituitary disease
• Functional gonadotrophin deficiency
Chronic systemic illness (e.g. asthma, malabsorption, coeliac disease, cystic fibrosis, renal failure)
Psychological stress
Anorexia nervosa
Excessive physical exercise
Hyperprolactinaemia
Other endocrine disease (e.g. Cushing’s syndrome,
primary hypothyroidism)
• Isolated gonadotrophin deficiency (Kallmann’s syndrome)
Hypergonadotrophic hypogonadism
• Acquired gonadal damage
Chemotherapy/radiotherapy to gonads .Trauma/surgery to gonads . Autoimmune gonadal failure . Mumps orchitis.
Tuberculosis. Haemochromatosis
• Developmental/congenital gonadal disorders.
Steroid biosynthetic defects
Anorchidism/cryptorchidism in males. Klinefelter’s syndrome (47XXY, male phenotype).
Turner’s syndrome (45XO, female phenotype)

Delayed puberty
Constitutional delay of pubertyThis is the most common cause of delayed puberty.
Affected children are healthy and have usually been
more than 2 SD below the mean height for their age
throughout childhood. There is often a history of delayed
puberty in siblings or parents. Since sex steroids are
essential for fusion of the epiphyses, ‘bone age’ can be
estimated by X-rays of epiphyses, usually in the wrist
and hand; in constitutional delay, bone age is lower
than chronological age. Constitutional delay of puberty
should be considered as a normal variant, as puberty
will commence spontaneously. However, affected children
can experience significant psychological distress
because of their lack of physical development, particularly
when compared with their peers.
Hypogonadotrophic hypogonadism
This may be due to structural, inflammatory or infiltrative
disorders of the pituitary and/or hypothalamus. In such circumstances, other pituitary hormones, such as growth hormone, are also likely to be deficient.
‘Functional’ gonadotrophin deficiency is caused by a
variety of factors, including low body weight, chronic
systemic illness (as a consequence of the disease itself or
secondary malnutrition), endocrine disorders and profound
psychosocial stress.
Isolated gonadotrophin deficiency is usually due to a
genetic abnormality that affects the synthesis of eitherGnRH or gonadotrophins. The most common form is
Kallmann’s syndrome, in which there is primary GnRH
deficiency and, in most affected individuals, agenesis or
hypoplasia of the olfactory bulbs, resulting in anosmia
or hyposmia. If isolated gonadotrophin deficiency is left
untreated, the epiphyses fail to fuse, resulting in tall
stature with disproportionately long arms and legs
relative to trunk height (eunuchoid habitus).
Cryptorchidism (undescended testes) and gynaecomastia
are commonly observed in all forms of hypogonadotrophic
hypogonadism.
Hypergonadotrophic hypogonadism
Hypergonadotrophic hypogonadism associated with
delayed puberty is usually due to Klinefelter’s syndrome
in boys and Turner’s syndrome in girls .
Other causes of primary gonadal failure are shown in Box .
Investigations
Key measurements are LH and FSH, testosterone (in boys) and oestradiol (in girls). Chromosome analysis should be performed if gonadotrophin concentrations are elevated. If gonadotrophin concentrations are low, then the differential diagnosis lies between constitutional delay and hypogonadotrophic hypogonadism. A plain X-ray of the wrist and hand may be compared with a set of standard films to obtain a bone age. Full blood count, renal function, liver function, thyroid function and coeliac disease autoantibodies should be measured, but further tests may be unnecessary if the blood tests are normal and the child has all the clinical features of constitutional delay. If hypogonadotrophic hypogonadism is suspected, neuroimaging and further investigations are required .Management
Puberty can be induced using low doses of oral oestrogen in girls (for example, ethinylestradiol 2 μg daily) or testosterone in boys (testosterone gel or depot testosterone esters). Higher doses carry a risk of early fusion of epiphyses. This therapy should be given in a specialist clinic where the progress of puberty and growth can be carefully monitored. In children with constitutionaldelay, this ‘priming’ therapy can be discontinued when
endogenous puberty is established, usually in less than a
year. In children with hypogonadism, the underlying
cause should be treated and reversed if possible. If
hypogonadism is permanent, sex hormone doses are
gradually increased during puberty and full adult
replacement doses given when development is complete.
Amenorrhoea
Primary amenorrhoea describes the condition of afemale patient who has never menstruated; this usually
occurs as a manifestation of delayed puberty but may
also be a consequence of anatomical defects of the female
reproductive system, such as endometrial hypoplasia or
vaginal agenesis.
Secondary amenorrhoea describes the cessation of menstruation. The causes of this common presentation are shown in Box.
In non-pregnant women, secondary amenorrhoea is almost invariably a consequence of either ovarian or hypothalamic/pituitary dysfunction.
Premature ovarian failure (premature menopause) is defined, arbitrarily, as occurring before 40 years of age. Rarely, endometrial adhesions (Asherman’s
syndrome) can form after uterine curettage,
surgery or infection with tuberculosis or schistosomiasis,
preventing endometrial proliferation and shedding.
Causes of secondary amenorrhoea
Physiological• Pregnancy
• Menopause
Hypogonadotrophic hypogonadism
Ovarian dysfunction
• Hypergonadotrophic hypogonadism
• Polycystic ovarian syndrome
• Androgen-secreting tumours
Uterine dysfunction
• Asherman’s syndrome
Clinical assessment
The underlying cause can often be suspected fromassociated clinical features and the patient’s age.
Hypothalamic/pituitary disease and premature ovarian
failure result in oestrogen deficiency, which causes a
variety of symptoms usually associated with the menopause.
A history of galactorrhoea should be sought. Significant weight loss of any cause can cause amenorrhoea by suppression of gonadotrophins. Weight gain may suggest hypothyroidism, Cushing’s syndrome or, very rarely, a hypothalamic lesion. Hirsutism, obesity and long-standing irregular periods suggest polycystic ovarian syndrome .The presence of other autoimmune disease raises the possibility of autoimmune premature ovarian failure.
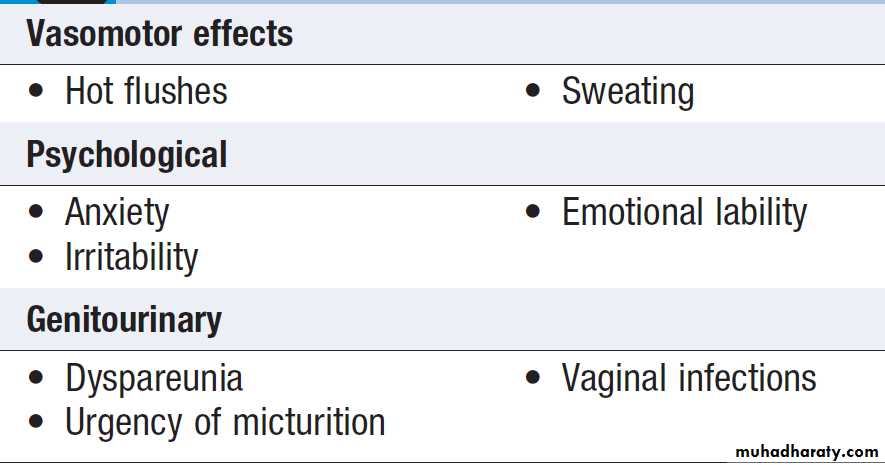
Symptoms of oestrogen deficiency
InvestigationsPregnancy should be excluded in women of reproductive
age by measuring urine or serum human chorionic
gonadotrophin (hCG). Serum LH, FSH, oestradiol, prolactin,
testosterone, T4 and TSH should be measured and, in the absence of a menstrual cycle, can be taken at any time. High concentrations of LH and FSH with low or low-normal oestradiol suggest primary ovarian failure. Ovarian autoantibodies may be positive when there is an underlying autoimmune aetiology, and a karyotype should be performed in younger women to exclude mosaic Turner’s syndrome. Elevated LH, prolactin and testosterone levels with normal oestradiol are common in PCOS.
Low levels of LH, FSH and oestradiol suggest hypothalamic or pituitary disease, and a pituitary MRI is indicated.
There is some overlap in gonadotrophin and oestrogen
concentrations between women with hypogonadotrophic
hypogonadism and PCOS. If there is doubt as to
the underlying cause of secondary amenorrhoea, then
the response to 5 days of treatment with an oral progestogen (e.g. medroxyprogesterone acetate 10 mg twice daily) can be assessed.
In women with PCOS, the progestogen will cause maturation of the endometrium and menstruation will occur a few days after the progestogen is stopped.
In women with hypogonadotrophic hypogonadism, menstruation does not occur following progestogen withdrawal because the endometrium is atrophic as a result of oestrogen deficiency. If doubt persists in distinguishing oestrogen deficiency from a uterine abnormality, the capacity for menstruation can be tested with 1 month of treatment with cyclical oestrogen and progestogen (usually administered as a combined oral contraceptive pill).
Assessment of bone mineral density by dual energy
X-ray absorptiometry (DEXA) may be appropriate
in patients with low androgen and oestrogen levels.
Management
Where possible, the underlying cause should be treated.
For example, women with functional amenorrhoea due
to excessive exercise and low weight should be encouraged
to reduce their exercise and regain some weight.
In oestrogen-deficient women, replacement therapy
may be necessary to treat symptoms and/or to prevent
osteoporosis. Women who have had a hysterectomy can
be treated with oestrogen alone, but those with a uterus
should be treated with combined oestrogen/ progestogen
therapy, since unopposed oestrogen increases the risk of endometrial cancer.
Cyclical hormone replacement therapy (HRT) regimens typically involve giving oestrogen on days 1–21 and progestogen on days 14–21 of the cycle and this can be conveniently administered as the oral contraceptive pill.
If oestrogenic side-effects (fluid retention, weight gain, hypertension and thrombosis) are a concern, then lower-dose oral or transdermal HRT may be more appropriate.
The timing of the discontinuation of oestrogen
replacement therapy is still a matter of debate.
In postmenopausal women, HRT has been shown to relieve
menopausal symptoms and to prevent osteoporotic fractures but is associated with adverse effects, which arerelated to the duration of therapy and to the patient’s
age (Box). In patients with premature menopause,
HRT should be continued up to the age of around
50 years, but only continued beyond this age if there
are continued symptoms of oestrogen deficiency on
discontinuation.

Hormone replacement therapy (HRT) in
post-menopausal womenMale hypogonadism
The clinical features of both hypo- and hypergonadotrophichypogonadism include loss of libido, lethargy
with muscle weakness, and decreased frequency of
shaving.
Patients may also present with gynaecomastia, infertility, delayed puberty, osteoporosis or anaemia of chronic disease.
Investigations
Male hypogonadism is confirmed by demonstrating alow serum testosterone level. The distinction between
hypo- and hypergonadotrophic hypogonadism is by
measurement of random LH and FSH.
Biochemical hypogonadism is associated with central
obesity and the metabolic syndrome postulated
mechanisms are complex and include reduction in sex
hormone binding globulin by insulin resistance and
reduction in GnRH and gonadotrophin secretion
by cytokines or oestrogen released by adipose tissue.
Testosterone levels also fall gradually with age
in men and this is associated with gonadotrophin levels that are low or inappropriately within the ‘normal’ range. There is an increasing trend to measure testosterone in older men, typically as part of an assessment of erectile dysfunction. Patients with hypergonadotrophic hypogonadism should have the testes examined for cryptorchidism or atrophy, and a karyotype performed
(to identify Klinefelter’s syndrome).
Management
Testosterone replacement is clearly indicated in youngermen with significant hypogonadism to prevent osteoporosis and to restore muscle power and libido.
Debate exists as to whether replacement therapy is of
benefit in mild hypogonadism associated with ageing
and central obesity, particularly in the absence of structural
pituitary/hypothalamic disease or other pituitary hormone deficiency. In such instances, a therapeutic trial of testosterone therapy may be considered if symptoms are present, but the benefits of therapy must be carefully weighed against the potential for harm. First-pass hepatic metabolism of testosterone is highly efficient, so bioavailability of ingested preparations is poor.
Doses of systemic testosterone can be titrated against symptoms; circulating testosterone levels may provide only a rough guide to dosage because they may be highly variable .
Testosterone therapy can aggravate prostatic carcinoma;
prostate-specific antigen (PSA) should be measured
before commencing testosterone therapy in men older
than 50 years and monitored annually thereafter.
Haemoglobin concentration should also be monitored in
older men, as androgen replacement can cause polycythaemia.
Testosterone replacement inhibits spermatogenesis;
treatment for fertility is described below.
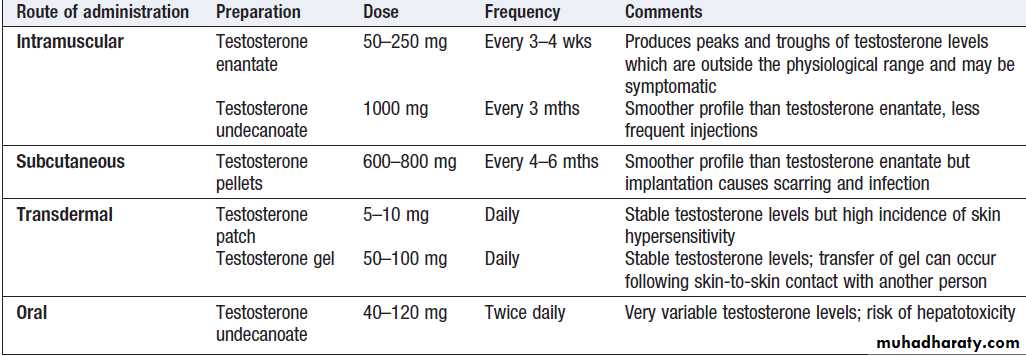
Options for androgen replacement therapy
InfertilityInfertility affects around 1 in 7 couples of reproductive
age, often causing psychological distress. In women, it may result from anovulation or abnormalities of the reproductive tract that prevent fertilisation or embryonic implantation,often damaged fallopian tubes from previous infection. In men, infertility may result from impaired sperm quality (for example, reduced motility) or reduced sperm number. Azoospermia or oligospermia is usually idiopathic, but may be a consequence of hypogonadism . Microdeletions of the Y chromosome are increasingly recognised as a cause of severely abnormal spermatogenesis. In many couples, more than one factor causing subfertility is present, and in a large proportion no cause can be identified.
Causes of infertility
Female factor (35–40%)• Ovulatory dysfunction
Polycystic ovarian syndrome
Hypogonadotrophic hypogonadism Hypergonadotrophic hypogonadism
• Tubular dysfunction
Pelvic inflammatory disease (chlamydia, gonorrhoea)
Endometriosis
Previous sterilisation
Previous pelvic or abdominal surgery
• Cervical and/or uterine dysfunction
Congenital abnormalities
Fibroids
Treatment for cervical carcinoma
Asherman’s syndrome
Male factor (35–40%)
• Reduced sperm quality or production Y chromosome microdeletions
Varicocoele
Hypergonadotrophic hypogonadism (see Hypogonadotrophic hypogonadism
• Tubular dysfunction
Varicocoele
Congenital abnormality of vas deferens/epididymis
Previous sexually transmitted infection (chlamydia,gonorrhoea)
Previous vasectomy
Unexplained or mixed factor
(20–35%)
Clinical assessment
A history of previous pregnancies, relevant infections
and surgery is important in both men and women. A
sexual history must be explored sensitively, as some
couples have intercourse infrequently or only when they
consider the woman to be ovulating, and psychosexual
difficulties are common. Irregular and/or infrequent
menstrual periods are an indicator of anovulatory cycles
in the woman, in which case causes such as PCOS should be considered. In men, the testes should be examined to
confirm that both are in the scrotum and to identify any
structural abnormality, such as small size, absent vas
deferens or the presence of a varicocoele.
Investigations
Investigations should generally be performed after acouple has failed to conceive despite unprotected intercourse for 12 months, unless there is an obvious abnormality like amenorrhoea. Both partners need to be
investigated. The male partner needs a semen analysis
to assess sperm count and quality. Home testing for
ovulation (by commercial urine dipstick kits, temperature
measurement, or assessment of cervical mucus) is
not recommended, as the information is often counterbalanced by increased anxiety if interpretation is inconclusive. In women with regular periods, ovulation can be confirmed by an elevated serum progesterone concentration on day 21 of the menstrual cycle.
Transvaginal ultrasound can be used to assess uterine and ovarian anatomy. Tubal patency may be examined at laparoscopy or by hysterosalpingography (HSG; a radio-opaque medium is injected into the uterus and should normally outline the fallopian tubes).
In vitro assessments of sperm survival in cervical mucus may be done in cases of unexplained infertility but are rarely helpful.
Management
Couples should be advised to have regular sexual intercourse, ideally every 2–3 days throughout the menstrual cycle. It is not uncommon for ‘spontaneous’ pregnancies to occur in couples undergoing investigations for infertility or with identified causes of male or femalesubfertility. In women with anovulatory cycles secondary to
PCOS , clomifene, which has partial antioestrogen
action, blocks negative feedback of oestrogen on the hypothalamus/pituitary, causing gonadotrophin secretion and thus ovulation. In women with gonadotrophin deficiency or in whom anti-oestrogen therapy is unsuccessful, ovulation may be induced by direct stimulation of the ovary by daily injection of FSH and an injection of hCG to induce follicular rupture at the appropriate time.
In hypothalamic disease, pulsatile GnRH therapy with a portable infusion pump can be used to stimulate pituitary gonadotrophin secretion (note that non-pulsatile administration of GnRH or its analogues paradoxically suppresses LH and FSH secretion). Whatever method of ovulation induction is employed, monitoring of response is essential to avoid multiple ovulation. For clomifene, ultrasound monitoring is recommended for at least the first cycle. During gonadotrophin therapy, closer monitoring of follicular growth by transvaginal ultrasonography and blood oestradiol levels is mandatory. ‘Ovarian hyperstimulation syndrome’ is characterised by grossly enlarged ovaries and capillary leak with circulatory shock, pleural effusions and ascites.
Anovulatory women who fail to respond to ovulation induction or who have primary ovarian failure may wish to consider using donated eggs or embryos, surrogacy and adoption.
Surgery to restore fallopian tube patency can be effective
but in vitro fertilisation (IVF) is normally recommended.
IVF is widely used for many causes of infertility and in unexplained cases of prolonged (> 3 years) infertility. The success of IVF depends on age, with low success rates in women over 40 years.
Men with hypogonadotrophic hypogonadism who
wish fertility are usually given injections of hCG severaltimes a week (recombinant FSH may also be required in
men with hypogonadism of pre-pubertal origin); it may
take up to 2 years to achieve satisfactory sperm counts.
Surgery is rarely an option in primary testicular disease
but removal of a varicocoele can improve semen quality.
Extraction of sperm from the epididymis for IVF, and
intracytoplasmic sperm injection (ICSI, when single
spermatozoa are injected into each oِcyte) are being
used increasingly in men with oligospermia or poor
sperm quality who have primary testicular disease.
Azoospermic men may opt to use donated sperm but
this may be in short supply.
Gynaecomastia
Gynaecomastia is the presence of glandular breast tissue
in males. Normal breast development in women is
oestrogen-dependent, while androgens oppose this
effect. Gynaecomastia results from an imbalance between androgen and oestrogen activity, which may reflect androgen deficiency or oestrogen excess.
The most common are physiological:
in the newborn baby (due to maternal and placental oestrogens), in pubertal boys (in whom oestradiolconcentrations reach adult levels before testosterone)
and in elderly men (due to decreasing testosterone).
Prolactin excess alone does not cause gynaecomastia .
Causes of gynaecomastia
IdiopathicPhysiological
Drug-induced
• Cimetidine
• Digoxin
• Anti-androgens (cyproterone acetate, spironolactone)
• Some exogenous anabolic steroids (diethylstilbestrol)
• Cannabis
Hypogonadism
Androgen resistance syndromes
Oestrogen excess
• Liver failure (impaired steroid metabolism)
• Oestrogen-secreting tumour (for example, of testis)
• hCG-secreting tumour (for example, of testis or lung)
Clinical assessment
A drug history is important. Gynaecomastia is often
asymmetrical and palpation may allow breast tissue to
be distinguished from the prominent adipose tissue
around the nipple that is often observed in obesity. Features of hypogonadism should be sought (see above)
and the testes examined for evidence of cryptorchidism,
atrophy or a tumour.
Investigations
If a clinical distinction between gynaecomastia and
adipose tissue cannot be made, then ultrasonography or
mammography is required. A random blood sample should be taken for testosterone, LH, FSH, oestradiol, prolactin and hCG. Elevated oestrogen concentrations are found in testicular tumours and hCG-producing neoplasms.
Management
An adolescent with gynaecomastia who is progressingnormally through puberty may be reassured that the
gynaecomastia will usually resolve once development is
complete. If puberty does not proceed in a harmonious
manner, then there may be an underlying abnormality
that requires investigation . Gynaecomastia may
cause significant psychological distress, especially in
adolescent boys, and surgical excision may be justified
for cosmetic reasons. Androgen replacement will usually
improve gynaecomastia in hypogonadal males and any
other identifiable underlying cause should be addressed
if possible. The anti-oestrogen tamoxifen may also be
effective in reducing the size of the breast tissue.
Hirsutism
Hirsutism refers to the excessive growth of thick terminal
hair in an androgen-dependent distribution in
women (upper lip, chin, chest, back, lower abdomen,
thigh, forearm) and is one of the most common presentations
of endocrine disease.
It should be distinguished from
Hypertrichosis,
which is generalised excessive growth of vellus hair.
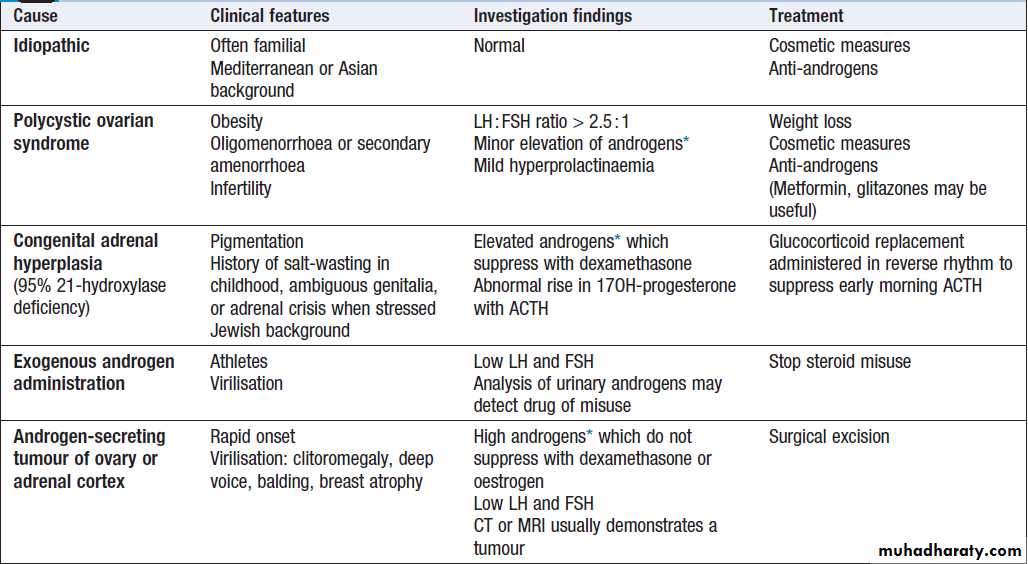


Causes of hirsutism
Clinical assessment
The severity of hirsutism is subjective. Some women suffer profound embarrassment from a degree of hair growth which others would not consider remarkable.
Important observations are a drug and menstrual history, calculation of body mass index, measurement of blood pressure, and examination for virilisation (clitoromegaly, deep voice, male-pattern balding, breast atrophy) and associated features, including acne vulgaris or Cushing’s syndrome.
Hirsutism of recent onset associated with virilisation is suggestive of an androgen-secreting tumour but this is rare.
Investigations
A random blood sample should be taken for testosterone,prolactin, LH and FSH. If there are clinical features
of Cushing’s syndrome, further investigations should be
performed .
If testosterone levels are more than twice the upper limit of normal for females, idiopathic hirsutism and PCOS are less likely, especially if LH and FSH levels are low. Under these circumstances, other causes of androgen excess should be sought. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is diagnosed by a short ACTH stimulation test with measurement of 17OH-progesterone .
In patients with androgen- secreting tumours, serum testosterone does not suppress following dexamethasone (either as an overnight or a 48-hour low-dose suppression test) or oestrogen (30 μg daily for 7 days). The tumour should then be sought by CT or MRI of the adrenals and ovaries.
Management
This depends on the cause . Options for
the treatment of PCOS and idiopathic hirsutism are
similar and are described below.
Polycystic ovarian syndrome (PCOS)
Affects up to 10% of women of reproductive age. It is a heterogenous disorder often associated with obesity, for which the primary cause remains uncertain. Geneticfactors probably play a role, since PCOS often affects
several family members. The severity and clinical features
vary markedly but diagnosis is usually made during the investigation of hirsutism or amenorrhoea/ oligomenorrhoea. Infertility may also be present.
Diagnosis of PCOS requires the presence of two of the following three features:
• menstrual irregularity
• clinical or biochemical androgen excess
• multiple cysts in the ovaries (most readily
detected by transvaginal ultrasound.
Polycystic ovary.
A transvaginal ultrasound scan showingmultiple cysts (some indicated by small arrows) in the ovary (highlighted by
bigger arrows) of a woman with polycystic ovarian syndrome.
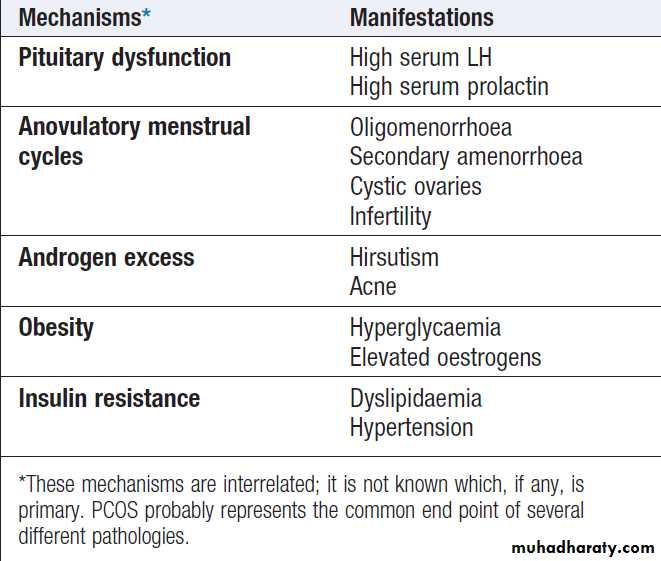
Features of polycystic ovarian syndrome
Women with PCOS are at increased risk of glucoseintolerance and some authorities recommend screening
for type 2 diabetes and other cardiovascular risk factors
associated with the metabolic syndrome .
Management
This should be directed at the presenting complaint, but all PCOS patients who are overweight should be encouraged to lose weight, as this can improve several symptoms, including menstrual irregularity, and reduces the risk of type 2 diabetes.
Menstrual irregularity and infertility
Most women with PCOS have oligomenorrhoea, with
irregular, heavy menstrual periods. This may not require
treatment unless fertility is desired.
Metformin , by reducing insulin resistance, may restore regular ovulatory cycles in overweight women, although it is less effective than clomifene at restoring fertility as measured by successful pregnancy .Thiazolidinediones also enhance insulin sensitivity and restore menstrual regularity in PCOS, but are contraindicated in women planning pregnancy. In women who have very few periods each year or are amenorrhoeic, the high oestrogen concentrations associated with PCOS can cause endometrial hyperplasia. Progestogens can be administered on a cyclical basis to induce regular shedding of the endometrium and a withdrawal bleed, or a progestogen-impregnated intrauterine coil can be fitted.
Hirsutism
Most patients will have used cosmetic measures, such as shaving, bleaching and waxing, before consulting a doctor. Electrolysis and laser treatment are effective for small areas like the upper lip and for chest hair but are expensive. Eflornithine cream inhibits ornithine decarboxylase in hair follicles and may reduce hair growth when applied daily to affected areas of the face. If conservative measures are unsuccessful, antiandrogen therapy is given . The life cycle of a hair follicle is at least 3 months and no improvement is likely before this time, when follicles have shed their hair and replacement hair growth has been suppressed. Metformin and thiazolidinediones are less effective at treating hirsutism than at restoring menstrual regularity. Unless weight is lost, hirsutism will return if therapy is discontinued.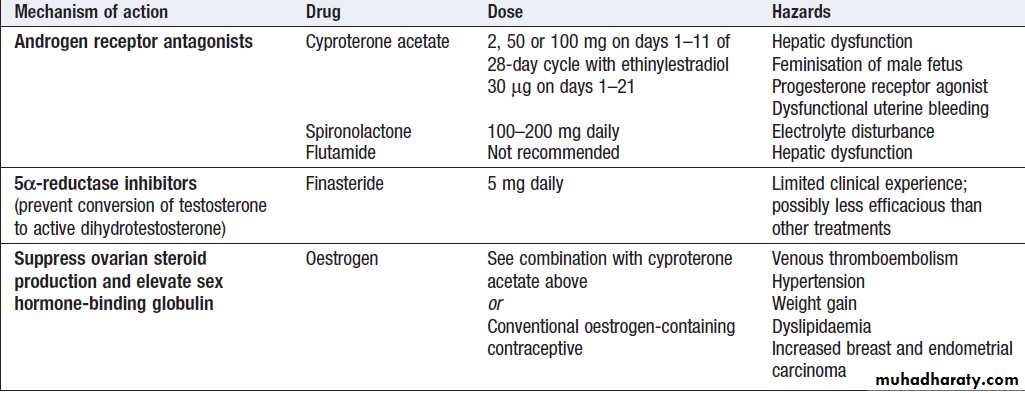
Anti-androgen therapy

Treatment of infertility in women with PCOS
Turner’s syndromeTurner’s syndrome affects around 1 in 2500 females. It
is classically associated with a 45XO karyotype but other
cytogenetic abnormalities may be responsible, including
mosaic forms (e.g. 45XO/46XX or 45XO/46XY) and
partial deletions of an X chromosome.
Clinical features
These are shown in Figure . Individuals with Turner’s syndrome invariably have short stature from an early age and this is often the initial presenting symptom. It is probably due to haploinsufficiency of the SHOX gene, one copy of which is found on both the X and Y chromosomes, which encodes a protein that is predominantly found in bone fibroblasts.
The genital tract and external genitalia in Turner’s syndrome are female in character, since this is the default developmental outcome in the absence of testes. Ovarian tissue develops normally until the third month of gestation, but thereafter there is gonadal dysgenesis with
accelerated degeneration of oِcytes and increased ovarian stromal fibrosis, resulting in ‘streak ovaries’.
The inability of ovarian tissue to produce oestrogen results in loss of negative feedback and elevation of FSH and LH concentrations. There is a wide variation in the spectrum of associated somatic abnormalities. The severity of the phenotype is, in part, related to the underlying cytogenetic abnormality. Mosaic individuals may have only mild short stature and may enter puberty spontaneously before developing gonadal failure.
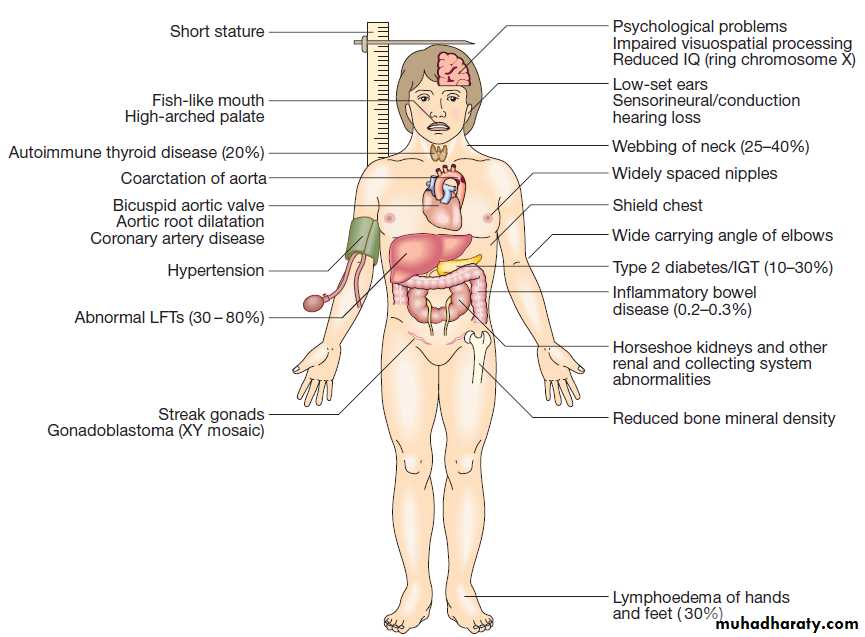
Clinical features of Turner’s syndrome (45XO).
(IGT = impaired glucose tolerance).Diagnosis and management
The diagnosis of Turner’s syndrome can be confirmedby karyotype analysis. Short stature, although not
directly due to growth hormone deficiency, responds to
high doses of growth hormone. Prophylactic gonadectomy
is recommended for individuals with 45XO/46XY mosaicism because there is an increased risk of gonadoblastoma. Pubertal development can be induced with oestrogen therapy but causes fusion of the epiphyses and cessation of growth. Therefore, the timing of pubertal induction needs to be carefully planned. Adults with Turner’s syndrome require long-term oestrogen replacement therapy and should be monitored periodically for the development of aortic root dilatation, hearing loss and other somatic complications.
Klinefelter’s syndrome
Klinefelter’s syndrome affects approximately 1 in1000 males and is usually associated with a 47XXY
karyotype. However, other cytogenetic variants may be responsible, especially 46XY/47XXY mosaicism. The
principal pathological abnormality is dysgenesis of the
seminiferous tubules. This is evident from infancy (and
possibly even in utero) and progresses with age. By adolescence, hyalinisation and fibrosis are present within
the seminiferous tubules and Leydig cell function is
impaired, resulting in hypogonadism.
Clinical features
The diagnosis is typically made in adolescents who have
presented with gynaecomastia and failure to progress
normally through puberty. Affected individuals usually
have small, firm testes. Tall stature is apparent from
early childhood, reflecting characteristically long leg
length associated with 47XXY, and may be exacerbated
by androgen deficiency with lack of epiphyseal closure
in puberty. Other clinical features may include learning
difficulties and behavioural disorders, as well as an
increased risk of breast cancer and type 2 diabetes in
later life. The spectrum of clinical features is wide and
some individuals, especially those with 46XY/47XXY
mosaicism, may pass through puberty normally and be
identified only during investigation for infertility.
Diagnosis and management
Klinefelter’s syndrome is suggested by the typical phenotype in a patient with hypergonadotrophic hypogonadism and can be confirmed by karyotype analysis. Individuals with clinical and biochemical evidence of androgen deficiency require androgen replacement .
Gonadal function in old age
THE PARATHYROID GLANDS
Parathyroid hormone (PTH) plays a key role in the regulation of calcium , phosphate and vitamin D metabolism.Functional anatomy, physiology and investigations
The four parathyroid glands lie behind the lobes of the thyroid and weigh between 25 and 40 mg. The parathyroid chief cells respond directly to changes in calcium via a G-protein-coupled cell surface receptor (the calcium-sensing receptor) located on the cell surface .When serum ionised calcium levels fall, PTH secretion rises. PTH is a single-chain polypeptide of 84 amino acids. It acts on the renal tubules to promote reabsorption of calcium and reduce reabsorption of phosphate, and on the skeleton to increase osteoclastic bone resorption and bone formation.
PTH promotes conversion of 25-hydroxycholecalciferol to the active metabolite 1,25-dihydroxycholecalciferol that enhances calcium absorption from the gut.
More than 99% of total body calcium is in bone. Prolonged exposure of bone to high levels of PTH is associated with increased osteoclastic activity and new bone formation, but the net effect is to cause bone loss with mobilisation of calcium into the extracellular fluid.
In contrast, pulsatile release of PTH causes net bone gain, an effect that is exploited therapeutically in the treatment of osteoporosis .
The differential diagnosis of disorders of calcium metabolism requires measurement of calcium phosphate, alkaline phosphatase, renal function, PTH and 25(OH)D.
Although the parathyroid glands detect and respond to ionised calcium levels, most clinical laboratories
only measure total serum calcium levels and about
50% of total calcium is bound to organic ions, such as
citrate or phosphate, and to proteins, especially albumin.
Accordingly, if the serum albumin level is reduced, total
calcium concentrations should be ‘corrected’ by adjusting
the value for calcium upwards by 0.02 mmol/L (0.4 mg/ dL) for each 1 g/L reduction in albumin below 40 g/L.
If albumin concentrations are significantly low,
as in severe acute illness and other chronic illness such
as liver cirrhosis, this correction is less accurate and
measurement of ionised calcium is needed.
Calcitonin is secreted from the parafollicular C cells
of the thyroid gland.
Although it is a useful tumour marker in medullary carcinoma of thyroid and can be given therapeutically in Paget’s disease of bone its release from the thyroid is of no clinical relevance to calcium homeostasis in humans.
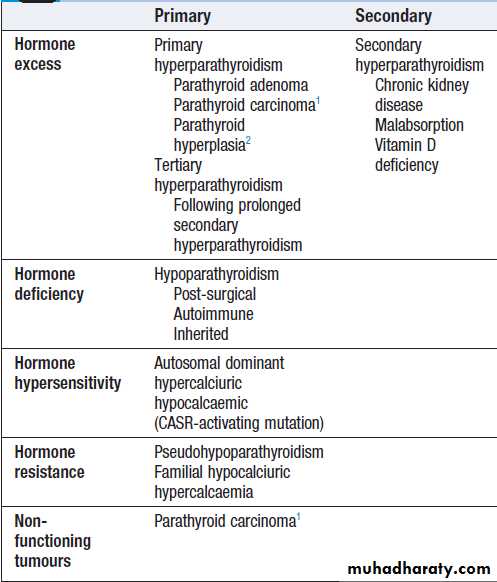
1Parathyroid carcinomas may or may not produce PTH. 2ln multiple
endocrine neoplasia (MEN) syndromes (CASR = calcium-sensing receptor)Classification of diseases of
the parathyroid glands
Presenting problems in parathyroid disease
HypercalcaemiaHypercalcaemia is one of the most common biochemical
abnormalities and is often detected during routine biochemical analysis in asymptomatic patients. However, it
can present with chronic symptoms, as described below,
and occasionally as an acute emergency with severe
hypercalcaemia and dehydration.
Of these, primary hyperparathyroidism (HPT) and malignant hypercalcaemia are by far the most common. Familial hypocalciuric hypercalcaemia (FHH) is a rare but important cause that needs differentiation from primary
HPT. Lithium may cause hyperparathyroidism by reducing the sensitivity of the calcium-sensing receptor.
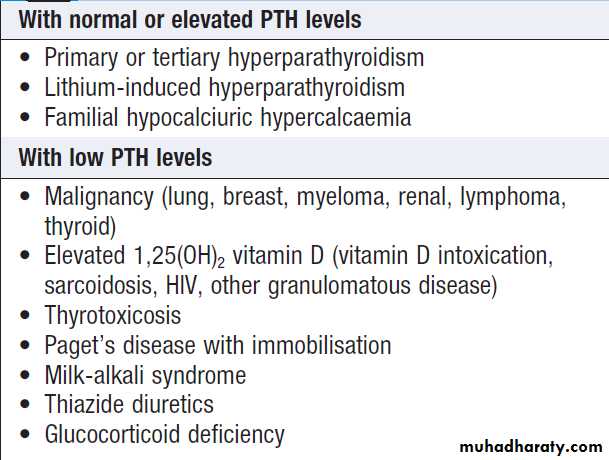
Causes of hypercalcaemia
Clinical assessmentSymptoms and signs of hypercalcaemia include polyuria
and polydipsia, renal colic, lethargy, anorexia,
nausea, dyspepsia and peptic ulceration, constipation,
depression, drowsiness and impaired cognition. Patients
with malignant hypercalcaemia can have a rapid onset
of symptoms and may have clinical features that help to
localise the tumour.
The classic symptoms of primary hyperparathyroidism
are described by the adage ‘bones, stones
and abdominal groans’, but few patients present in
this way nowadays and the disorder is most often picked
up as an incidental finding on biochemical testing.
About 50% of patients with primary hyperparathyroidism
are asymptomatic while others have nonspecificsymptoms such as fatigue, depression and generalised aches and pains.
Some present with renal calculi and it has been estimated that 5% of first stone formers and 15% of recurrent stone formers have primary hyperparathyroidism .
Hypertension is a common feature of hyperparathyroidism. Parathyroid tumours are almost never palpable.
A family history of hypercalcaemia raises the possibility
of FHH or MEN .
Investigations
The most discriminant investigation is measurement of
PTH. If PTH levels are detectable or elevated in the
presence of hypercalcaemia, then primary hyperparathyroidism is the most likely diagnosis. High plasma phosphate and alkaline phosphatase accompanied by renal impairment suggest tertiary hyperparathyroidism.
Hypercalcaemia may cause nephrocalcinosis and renal
tubular impairment, resulting in hyperuricaemia and
hyperchloraemia.
Patients with FHH can present with a similar biochemical picture to primary hyperparathyroidism but typically have low urinary calcium excretion (a ratio of urinary calcium clearance to creatinine clearance of < 0.01).
The diagnosis of FHH can be confirmed by screening family members for hypercalcaemia and/or
a mutation in the gene encoding the calcium-sensing
receptor. If PTH is low and no other cause is apparent, then
malignancy with or without bony metastases is likely.
PTH-related peptide, which is often responsible for the
hypercalcaemia associated with malignancy, is not
detected by PTH assays, but can be measured by a specific
assay (although this is not usually necessary).
Unless the source is obvious, the patient should be
screened for malignancy with a chest X-ray, myeloma
screen and CT as appropriate.
Management
Treatment of severe hypercalcaemia and primary hyperparathyroidism is described .
FHH does not require any specific intervention.
Hypocalcaemia
AetiologyMuch less common than hypercalcaemia. The most common cause is a low serum albumin with normal ionised calcium concentration. Conversely, ionised calcium may be low in the face of normal total serum calcium in patients with alkalosis: for example, as a result of hyperventilation.
Hypocalcaemia may also develop as a result of magnesium depletion and should be considered in patients with malabsorption, on diuretic or proton pump inhibitor and/or with a history of alcohol excess. Magnesium deficiency causes hypocalcaemia by impairing the ability of the parathyroid glands to secrete PTH (resulting in PTH concentrations that are low) and may also impair the actions of PTH on bone and kidney.

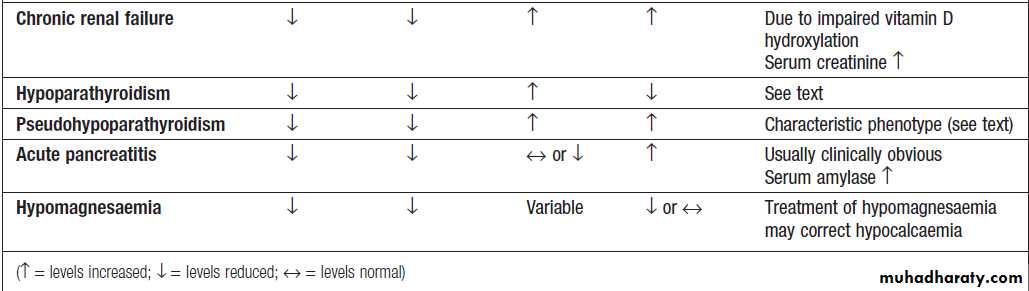

Differential diagnosis of hypocalcaemia
Clinical assessmentMild hypocalcaemia is often asymptomatic but, with
more profound reductions in serum calcium, tetany can
occur. This is characterised by muscle spasms due to
increased excitability of peripheral nerves.Children are more liable to develop tetany than adults and present with a characteristic triad of carpopedal spasm, stridor and convulsions, although one or more of these may be found independently of the others. In carpopedal spasm, the hands adopt a characteristic position with flexion of the metacarpophalangeal joints of the fingers and adduction of the thumb (‘main d’accoucheur’).
Pedal spasm can also occur but is less frequent. Stridor is caused by spasm of the glottis.
Adults can also develop carpopedal spasm in association
with tingling of the hands and feet and around the
mouth, but stridor and fits are rare.
Latent tetany may be detected by eliciting Trousseau’s
sign; inflation of a sphygmomanometer cuff on
the upper arm to more than the systolic blood pressure
is followed by carpal spasm within 3 minutes. Less specific
is Chvostek’s sign, in which tapping over the
branches of the facial nerve as they emerge from the
parotid gland produces twitching of the facial muscles.
Hypocalcaemia can cause papilloedema and prolongation
of the ECG QT interval, which may predisposeto ventricular arrhythmias. Prolonged hypocalcaemia
and hyperphosphataemia (as in hypoparathyroidism)
may cause calcification of the basal ganglia, grand mal
epilepsy, psychosis and cataracts. Hypocalcaemia associated with hypophosphataemia, as in vitamin D deficiency, causes rickets in children and osteomalacia in
adults .
Management
Emergency management of hypocalcaemia associated
with tetany is described in Box . Treatment of
chronic hypocalcaemia is described below .

Management of severe hypocalcaemia
Primary hyperparathyroidismCaused by autonomous secretion of PTH, usually by a single parathyroid adenoma, which can vary in diameter from a few millimetres to several centimetres. It should be distinguished from secondary hyperparathyroidism, in which there is a physiological increase in PTH secretion to compensate for prolonged hypocalcaemia (such as in vitamin D deficiency,), and from tertiary, in which continuous stimulation of the parathyroids over a prolonged period of time results in adenoma formation and autonomous PTH secretion . It is 2–3 times more common in women than men; 90% of patients are over 50 years of age. It also occurs in the familial MEN syndromes in which case hyperplasia or multiple adenomas of all four parathyroid glands are more likely than a solitary adenoma.
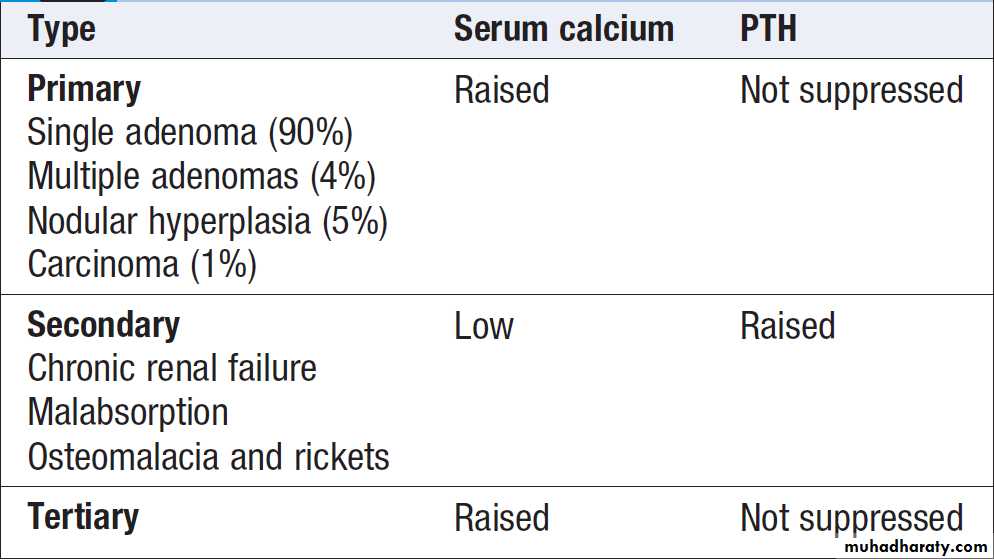
Hyperparathyroidism
Clinical and radiological featuresThe clinical presentation of primary hyperparathyroidism
is described. Parathyroid bone disease is now rare due to earlier diagnosis and treatment.
Osteitis fibrosa results from increased bone resorption by osteoclasts with fibrous replacement in the lacunae. This may present as bone pain and tenderness, fracture and deformity. Chondrocalcinosis can occur due to deposition of calcium pyrophosphate crystals within articular cartilage. It typically affects the menisci at the knees and can result in secondary degenerative arthritis or predispose to attacks of acute pseudogout.
Skeletal X-rays are usually normal in mild primary HPT but in patients with advanced disease characteristic changes are observed. In the early stages there is demineralisation, with subperiosteal erosions and terminal resorption in the phalanges. A ‘pepper-pot’ appearance may be seen on lateral X-rays of the skull. Reduced bone mineral density, resulting in either osteopenia or osteoporosis, is now the most common skeletal manifestation.
This is usually not evident radiographically and requires assessment by DEXA . In nephrocalcinosis, scattered opacities may be visible within the renal outline.
There may be soft tissue calcification in arterial walls and hands and in the cornea.
Investigations
The diagnosis can be confirmed by finding a raised PTH
level in the presence of hypercalcaemia, provided that
FHH is excluded. Parathyroid scanning by 99mTcsestamibi
scintigraphy and/or ultrasound examination can be performed prior to surgery, in an attempt to localise an adenoma and allow a targeted resection. However, negative imaging does not exclude the diagnosis.
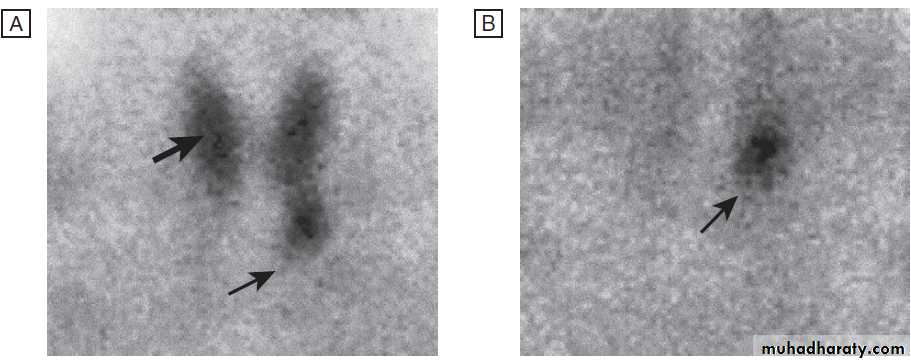
99mTc-sestamibi scan - a patient with primary hyperparathyroidism
secondary to a parathyroid adenoma.A After 1 hour, there is uptake in the thyroid gland (thick arrow) and the enlarged left inferior parathyroid gland (thin arrow).
B After 3 hours, uptake is evident only in the parathyroid.
Management
The treatment of choice for primary hyperparathyroidismis surgery, with excision of a solitary parathyroid adenoma or hyperplastic glands. Experienced surgeons will identify solitary tumours in more than 90% of cases. Patients with parathyroid bone disease run a significant risk of developing hypocalcaemia postoperatively, but the risk of this can be reduced by correcting vitamin D deficiency pre-operatively. Surgery is usually indicated for individuals aged < 50 years, with clear-cut symptoms or documented complications (such as peptic ulceration, renal stones, renal impairment or osteoporosis), and (in asymptomatic patients) significant hypercalcaemia (corrected serum calcium > 2.85 mmol/L (> 11.4 mg/dL)).
Patients who are treated conservatively should have calcium biochemistry and renal function checked annually and bone density monitored. They should be encouraged to maintain a high oral fluid intake to avoid renal stones. Occasionally, primary HPT presents with severe life-threatening hypercalcaemia. This is often due to dehydration and should be managed with IV fluids and bisphosphonates. If this is not effective, then urgent parathyroidectomy should be considered.
Cinacalcet is a calcimimetic which enhances the sensitivity of the calcium-sensing receptor, so reducing PTH levels, and is licensed for tertiary hyperparathyroidism and as a treatment for patients with primary HPT who are unwilling to have surgery or are medically unfit.
Familial hypocalciuric hypercalcaemia
This autosomal dominant disorder is caused by an
inactivating mutation in one of the alleles of the calcium-sensing receptor gene, which reduces the ability of the
parathyroid gland to ‘sense’ ionised calcium.
As a result, higher than normal calcium levels are
required to suppress PTH secretion. The typical presentation
is with mild hypercalcaemia with PTH concentrations
that are ‘inappropriately’ at the upper end of the
reference range or are slightly elevated. Calcium-sensing
receptors in the renal tubules are also affected and this
leads to increased renal tubular reabsorption of calcium
and hypocalciuria. The hypercalcaemia of FHH is always
asymptomatic and complications do not occur.
The main risk of FHH is of the patient being subjected to an unnecessary (and ineffective) parathyroidectomy if misdiagnosed as having primary hyperparathyroidism. Testing of family members for hypercalcaemia is helpful in confirming the diagnosis and it is also possible to perform genetic testing. No treatment is necessary.
Hypoparathyroidism
The most common cause of hypoparathyroidism isdamage to the parathyroid glands (or their blood supply)
during thyroid surgery; post-operative hypocalcaemia
develops in 5.5% of patients overall but 9% of patients
undergoing total thyroidectomy. Rarely, hypoparathyroidism can occur as a result of infiltration of the glands with iron in haemochromatosis) or copper in Wilson’s disease .
There are a number of rare congenital or inherited
forms of hypoparathyroidism. One form is associated
with autoimmune polyendocrine syndrome type 1 and another with DiGeorge syndrome .
Autosomal dominant hypoparathyroidism (ADH) is
the mirror image of familial hypocalciuric hypercalcaemia
in that an activating mutation in the calcium-sensing receptor reduces PTH levels, resulting in hypocalcaemia and hypercalciuria.
Pseudohypoparathyroidism
In this disorder, the individual is functionally hypoparathyroid but, instead of PTH deficiency, there is tissue resistance to the effects of PTH, such that PTH concentrations are markedly elevated. The PTH receptor
itself is normal but the downstream signalling pathways
are defective due to mutations that affect GNAS1, which
encodes the Gsα protein, a molecule involved in signal
transduction downstream of the PTH receptor and
other G-protein-coupled receptors.
There are several subtypes but the most common (pseudohypoparathyroidism type 1a) is characterised by hypocalcaemia and hyperphosphataemia, in association with short stature, short fourth metacarpals and metatarsals,
rounded face, obesity and subcutaneous calcification;
these features are collectively referred to as Albright’s
hereditary osteodystrophy (AHO).
Type 1a pseudohypoparathyroidism
occurs only when the GNAS1 mutation is inherited on the maternal chromosome. The term pseudopseudohypoparathyroidism is used to describe patients who have clinical features of AHO but normal serum calcium and PTH concentrations; it occurs when the GNAS1 mutation is inherited on the paternal chromosome.
The inheritance of these disorders is an example of genetic imprinting .The difference in clinical features occurs as a result of the fact that renal cells exclusively express the maternal GNAS1 allele, whereas both maternal and paternal alleles are expressed in other cell types; this explains why maternal inheritance is associated with hypocalcaemia and resistance to PTH (which regulates serum calcium and phosphate levels largely by an effect on the renal tubule), and why paternal inheritance is associated with skeletal and other abnormalities in the absence of hypocalcaemia and raised PTH values.
Management of hypoparathyroidism
Persistent hypoparathyroidism and pseudohypoparathyroidism are treated with oral calcium salts and vitamin D analogues, either 1α-hydroxycholecalciferol (alfacalcidol) or 1,25-DHCC (calcitriol).
This therapy needs careful monitoring because of
the risks of iatrogenic hypercalcaemia, hypercalciuria
and nephrocalcinosis. Recombinant PTH is available
as subcutaneous injection therapy for osteoporosis
and, although not currently licensed, has been used in hypoparathyroidism (but not in pseudohypoparathyroidism).
It is much more expensive than calcium and vitamin D analogue therapy but has the advantage that it is less likely to cause hypercalciuria.
There is no specific treatment for AHO other than to try
to maintain calcium levels within the reference range
using active vitamin D metabolites.

The parathyroid glands in old age
THE ADRENAL GLANDSThe adrenals comprise several separate endocrine
glands within a single anatomical structure. The adrenal
medulla is an extension of the sympathetic nervous
system which secretes catecholamines into capillaries rather than synapses.
Most of the adrenal cortex is made up of cells which secrete cortisol and adrenal androgens, and form part of the hypothalamic–pituitary–adrenal (HPA) axis. The small outer glomerulosa of the cortex secretes aldosterone under the control of the renin– angiotensin system. These functions are important in the integrated control of cardiovascular, metabolic and immune responses to stress.
There is increasing evidence that subtle alterations in
adrenal function contribute to the pathogenesis of
common diseases such as hypertension, obesity and
type 2 diabetes mellitus.
Functional anatomy and physiology
Adrenal anatomy and function are shown in Figure.
Histologically, the cortex is divided into three
zones, but these function as two units (zona glomerulosa
and zonae fasciculata/ reticularis) which produce corticosteroids in response to humoral stimuli. Pathways
for the biosynthesis of corticosteroids are shown
in Figure. Investigation of adrenal function is
described under specific diseases below. The different
types of adrenal disease are shown in Box .
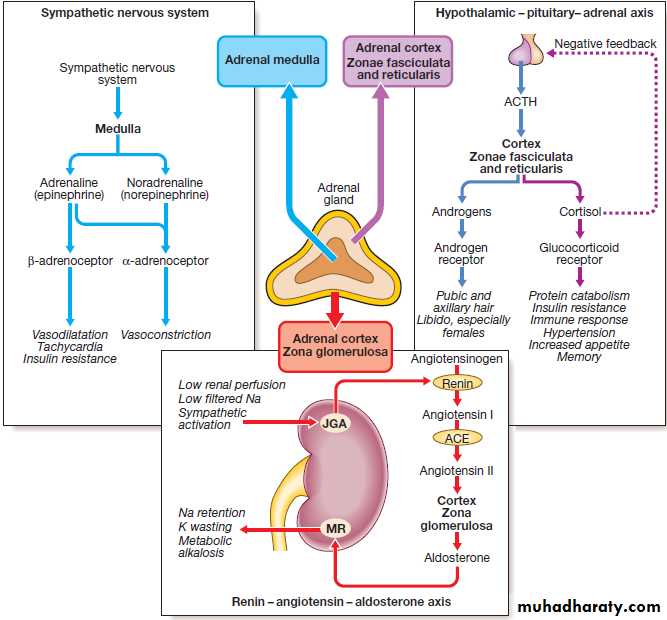
Structure and function of the adrenal glands. (ACE = angiotensin-converting enzyme; ACTH = adrenocorticotrophic hormone; JGA = juxtaglomerular apparatus; MR = mineralocorticoid receptor).
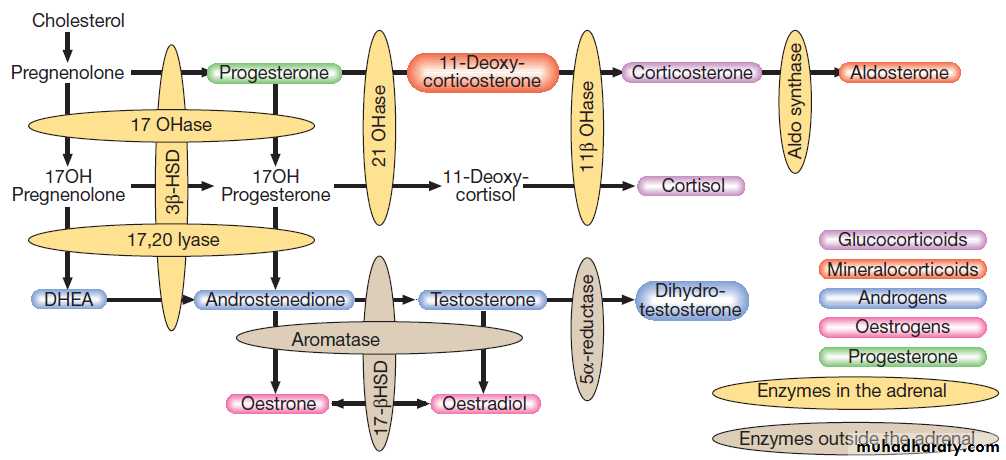
The major pathways of synthesis of steroid hormones. (DHEA = dehydroepiandrosterone; HSD = hydroxysteroid dehydrogenase;
OHase = hydroxylase)
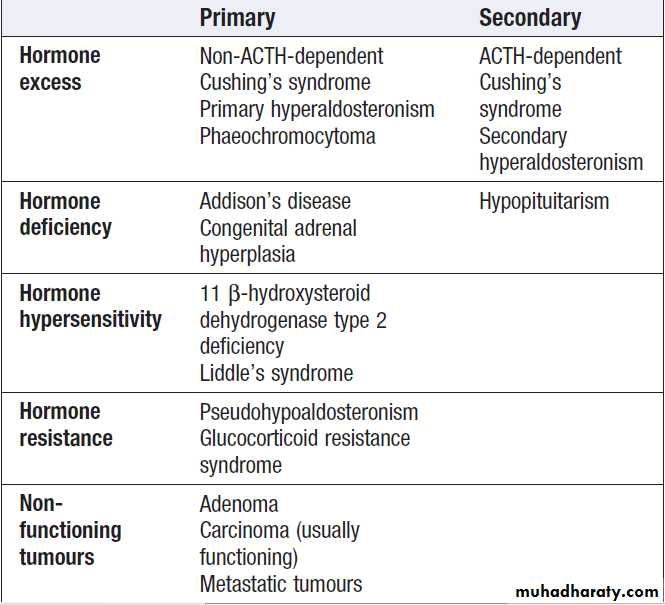
Classification of diseases of
the adrenal glandsGlucocorticoids
Cortisol is the major glucocorticoid in humans. Levelsare highest in the morning on waking and lowest in the
middle of the night. Cortisol rises dramatically during
stress, including any illness. This elevation protects key
metabolic functions (such as the maintenance of cerebral
glucose supply during starvation) and inhibits potentially
damaging inflammatory responses to infection
and injury. The clinical importance of cortisol deficiency
is, therefore, most obvious at times of stress.
More than 95% of circulating cortisol is bound to
protein, principally cortisol-binding globulin, which is increased by oestrogens. It is the free fraction that
is biologically active.
Cortisol regulates cell function by binding to glucocorticoid receptors that regulate the transcription of many genes. Cortisol can also activate mineralocorticoid receptors,
but it does not normally do so because most cells containing mineralocorticoid receptors also express an enzyme called 11 β-hydroxysteroid dehydrogenase type 2 (11 β-HSD2), which inactivates cortisol by converting it to cortisone.
Inhibitors of 11 β-HSD2 (such as liquorice) or mutations
in the gene that encodes 11 β-HSD2 cause cortisol to
act as a mineralocorticoid, resulting in sodium retention
and hypertension .
Mineralocorticoids
Aldosterone is the most important mineralocorticoid. Itbinds to mineralocorticoid receptors in the kidney and
causes sodium retention and increased excretion of
potassium and protons.The principal stimulus to aldosterone secretion is angiotensin II, a peptide produced by activation of the renin–angiotensin system . Renin activity in the juxtaglomerular apparatus of the kidney is stimulated by low perfusion pressure in the afferent arteriole, low sodium filtration leading to low sodium concentrations at the macula densa, or increased sympathetic nerve activity. As a result, renin activity is increased in hypovolaemia and
renal artery stenosis, and is approximately doubled
when standing up from a recumbent position.
Catecholamines
In humans, only a small proportion of circulatingnoradrenaline (norepinephrine) is derived from the
adrenal medulla; much more is released from sympathetic
nerve endings. Conversion of noradrenaline to
adrenaline (epinephrine) is catalysed by catechol- o-methyltransferase (COMT), which is induced by glucocorticoids.
Blood flow in the adrenal is centripetal, so that the medulla is bathed in high concentrations of cortisol and is the major source of circulating adrenaline.
However, after surgical removal of the adrenal medullae,
there appear to be no clinical consequences attributable
to deficiency of circulating catecholamines.
Adrenal androgens
Adrenal androgens are secreted in response to ACTH
and are the most abundant steroids in the blood stream.
They are probably important in the initiation of puberty
(adrenarche).
The adrenals are also the major source of androgens in adult females and may be important in female libido.
Presenting problems in adrenal disease
Cushing’s syndromeCushing’s syndrome is caused by excessive activation
of glucocorticoid receptors.
It is most commonly iatrogenic,
due to prolonged administration of synthetic
glucocorticoids such as prednisolone.
Endogenous Cushing’s syndrome is uncommon but is due to chronic over-production of cortisol by the adrenal glands, either as the result of an adrenal tumour or because of excessive production of ACTH by a pituitary tumour or
ectopic ACTH production by other tumours.
Aetiology
The causes are shown in Box. Amongst endogenouscauses, pituitary-dependent cortisol excess (by
convention, called Cushing’s disease) accounts for
approximately 80% of cases. Both Cushing’s disease and
cortisol-secreting adrenal tumours are four times more
common in women than men.
In contrast, ectopic ACTH syndrome (often due to a small-cell carcinoma of the bronchus) is more common in men.
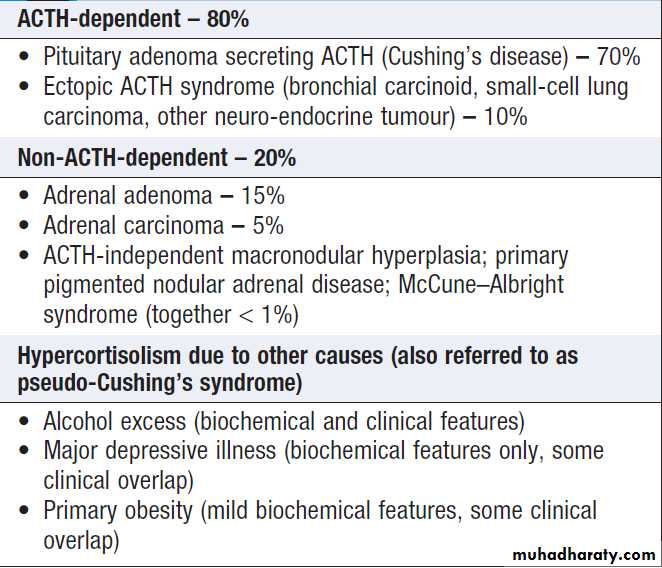
Classification of endogenous Cushing’s syndrome
Clinical assessmentThe diverse manifestations of glucocorticoid excess are
shown in Figure. Many of these are not specific to Cushing’s syndrome. Moreover, some common disorders can be confused with Cushing’s syndrome because they are associated with alterations in cortisol secretion: for example, obesity and depression . Features which favour Cushing’s syndrome in an obese patient are bruising, myopathy and thin skin. Any clinical suspicion of cortisol excess is best resolved by further investigation. It is vital to exclude iatrogenic causes in all patients with Cushing’s syndrome since even inhaled or topical glucocorticoids can induce the syndrome in susceptible individuals.
A careful drug history must therefore be taken before embarking on complex investigations. Some clinical features are more common in ectopic ACTH syndrome. Whilst ACTH-secreting pituitary tumours retain some negative feedback sensitivity to cortisol, this is absent in tumours that produce ectopic ACTH, typically resulting in higher levels of both ACTH and cortisol than are observed in pituitary-driven
disease. The high ACTH levels are associated with
marked pigmentation because of binding to melanocortin
1 receptors on melanocytes in the skin. The high
cortisol levels also overcome the capacity of 11 β-HSD2
to inactivate cortisol in the kidney, causing hypokalaemic alkalosis which aggravates myopathy and hyperglycaemia (by inhibiting insulin secretion).
When the tumour that is secreting ACTH is malignant,
then the onset is usually rapid and may be associatedwith cachexia. For these reasons, the classical features
of Cushing’s syndrome are less common in ectopic
ACTH syndrome; if present, they suggest that a less
aggressive tumour, such as a bronchial carcinoid, is
responsible.
In Cushing’s disease, the pituitary tumour is usually
a microadenoma (< 10 mm in diameter); features of a pituitary macroadenoma (hypopituitarism, visual failure or disconnection hyperprolactinaemia) are rare.
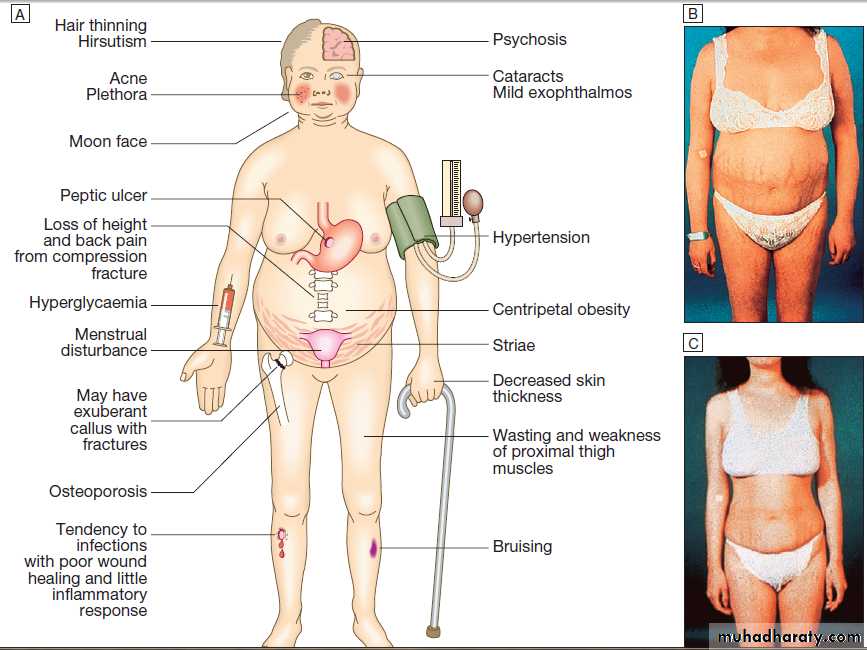
Fig- Cushing’s syndrome.
A Clinical features common to all causes.B A patient with Cushing’s disease before treatment.
C The same patient 1 year after the successful removal of an ACTH-secreting pituitary microadenoma by trans- sphenoidal surgery.
Investigations
The large number of tests available for Cushing’s syndromereflects the fact that each one has limited specificity
and sensitivity in isolation. Accordingly, several
tests are usually combined to establish the diagnosis.
Testing for Cushing’s syndrome should be avoided
under conditions of stress, such as an acute illness,
because this activates the HPA axis, causing potentially
spurious results.
The diagnosis of Cushing’s is a two-step process:
1. to establish whether the patient has Cushing’s
syndrome
2. to define its cause .
Some additional tests are useful in all cases of Cushing’s
syndrome, including plasma electrolytes, glucose,
glycosylated haemoglobin and bone mineral density
measurement.
Establishing the presence of Cushing’s syndrome
Cushing’s syndrome is confirmed by using two of three
main tests:
1. failure to suppress serum cortisol with low doses
of oral dexamethasone
2. loss of the normal circadian rhythm of cortisol, with inappropriately elevated late-night serum or salivary cortisol
3. increased 24-hour urine free cortisol
Dexamethasone is used for suppression testing
because it does not cross-react in radioimmunoassaysfor cortisol.
An overnight dexamethasone suppression test (ONDST) involves administration of 1 mg dexamethasone at 2300 hrs and measuring serum cortisol at 0900 hrs the following day.
In a low-dose dexamethasone suppression test (LDDST), serum cortisol is measured following administration of 0.5 mg dexamethasone 4 times daily for 48 hours.
It is important for any oestrogens to be stopped for 6 weeks prior to investigation to allow corticosteroid-binding globulin (CBG) levels to return to normal and to avoid false-positive responses, as most cortisol assays measure total cortisol, including that bound to CBG. Cyclicity of cortisol secretion is a feature of all types of Cushing’s syndrome and, if very variable, can confuse diagnosis. Use of multiple salivary cortisol samples over weeks or months
can be of use in diagnosis, but an elevated salivary
cortisol alone should not be taken as proof of diagnosis.
In iatrogenic Cushing’s syndrome, cortisol levels are
low unless the patient is taking a corticosteroid (such
as prednisolone) that cross-reacts in immunoassays
with cortisol.
Determining the underlying cause
Once the presence of Cushing’s syndrome is confirmed,
measurement of plasma ACTH is the key to establishing
the differential diagnosis. In the presence of excess
cortisol secretion, an undetectable ACTH (below
1.1 pmol/L (5 pg/mL)) indicates an adrenal cause,
while ACTH levels greater than 3.3 pmol/L (15 pg/mL)
suggest a pituitary cause or ectopic ACTH.
ACTH levels between these values represent a ‘grey area’, and further evaluation by a specialist is required. Tests to
discriminate pituitary from ectopic sources of ACTH
rely on the fact that pituitary tumours, but not ectopic
tumours, retain some features of normal regulation of
ACTH secretion. Thus, in pituitary-dependent Cushing’s
disease, ACTH secretion is suppressed by high dose
dexamethasone and ACTH is stimulated by
corticotrophin-releasing hormone (CRH). In a high-dose
dexamethasone suppression test (HDDST), serum cortisol
is measured before and after administration of 2 mg
of dexamethasone 4 times daily for 48 hours.
MRI detects around 60% of pituitary microadenomas secreting ACTH. If available, bilateral inferior petrosal sinus sampling (BIPSS) with measurement of ACTH is the best means of confirming Cushing’s disease, unless MRI
shows a tumour bigger than 6 mm.
CT or MRI detects most adrenal tumours; adrenal carcinomas are usually large (> 5 cm) and have other features of malignancy.
Sequence of investigations in suspected spontaneous Cushing’s syndrome. A serum cortisol of 50 nmol/L is equivalent to
1.81 μg/ dL. (LDDST = low-dose dexamethasone suppression test; ONDST = overnight dexamethasone suppression test; UFC = urinary free cortisol)
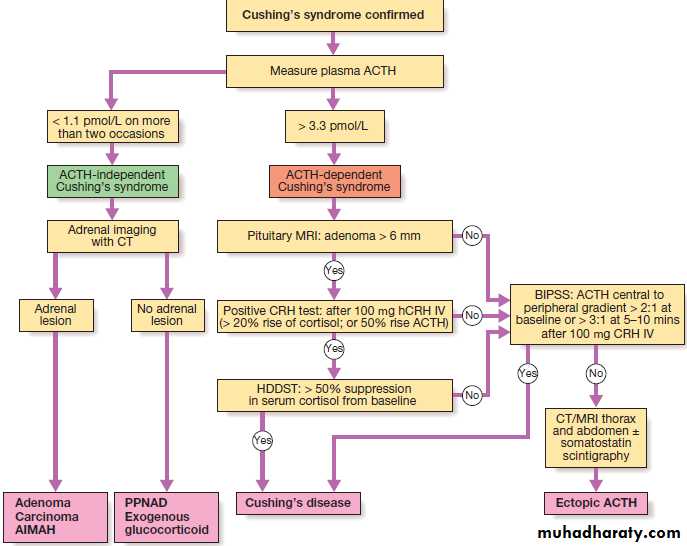
Determining the cause of confirmed Cushing’s syndrome. To convert pmol/L to ng/L, multiply by 4.541. (ACTH = adrenocorticotrophic
hormone; AIMAH = ACTH-independent macronodular adrenal hyperplasia; BIPSS = bilateral inferior petrosal sinus sampling; CRH = corticotrophin-releasing
hormone; HDDST = high-dose dexamethasone suppression test; PPNAD = primary pigmented nodular adrenal disease)
Management
Untreated severe Cushing’s syndrome has a 50% 5-yearmortality. Most patients are treated surgically, but
medical therapy may be given in severe cases for a few
weeks prior to operation to improve the clinical state.
A number of drugs are used to inhibit corticosteroid biosynthesis, including metyrapone and ketoconazole.
The dose of these agents is best titrated against serum cortisol levels or 24-hour urine free cortisol.
Cushing’s disease
Trans- sphenoidal surgery carried out by an experienced
surgeon with selective removal of the adenoma is the
treatment of choice, with approximately 70% of patients
going into immediate remission. Around 20% of patients
suffer a recurrence, often years later, emphasising the
need for life-long follow-up. Laparoscopic bilateral adrenalectomy performed by an expert surgeon effectively cures ACTH-dependent Cushing’s syndrome, but in patients with pituitary-dependent Cushing’s syndrome, this can result in Nelson’s syndrome, with an invasive pituitary macroadenoma and very high ACTH levels causing pigmentation. The risk of Nelson’s syndrome may be reduced by pituitary irradiation.
Adrenal tumours
Laparoscopic adrenal surgery is the treatment of choicefor adrenal adenomas. Surgery offers the only prospect
of cure for adrenocortical carcinomas, but in general,
prognosis is poor. Radiotherapy to the tumour bed reduces the risk of local recurrence, and systemic therapy consists of the adrenolytic drug mitotane and chemotherapy, but responses are often poor.
Ectopic ACTH syndrome
Localised tumours, such as bronchial carcinoid, should
be removed surgically. In patients with incurable malignancy, it is important to reduce the severity of the
Cushing’s syndrome using medical therapy (see above)
or, if appropriate, bilateral adrenalectomy.
Therapeutic use of glucocorticoids
The remarkable anti-inflammatory properties of glucocorticoids have led to their use in a wide variety of clinical conditions but the hazards are significant. Topical preparations (dermal, rectal and inhaled) can also be absorbed into the systemic circulation, and although this rarely occurs to a sufficient degree to produce clinical features of Cushing’s syndrome, it can result in significant suppression of endogenous ACTH and cortisol secretion. Severe Cushing’s syndrome can result if there is concomitant administration of inhaled glucocorticoids and inhibitors of the liver enzyme CYP450 3A4, such as the antiretroviral drug ritonavir .
Approximate equivalent doses of
glucocorticoidsAdverse effects of glucocorticoids
The clinical features of glucocorticoid excess are illustratedin Figure. Adverse effects are related to dose, duration of therapy, and pre-existing conditions that might be worsened by corticosteroid therapy, such as diabetes mellitus or osteoporosis. Osteoporosis is a particularly important problem because, for a given bone mineral density, the fracture risk is greater in glucocorticoid-treated patients than in post-menopausal osteoporosis. Therefore, when systemic glucocorticoids are prescribed and the anticipated duration of steroid therapy is more than 3 months, bone-protective therapy should be considered. Rapid changes in glucocorticoid levels can also lead to marked mood disturbances, including depression, mania and insomnia.
The anti-inflammatory effect of glucocorticoids may
mask signs of disease. For example, perforation of aviscus may be masked and the patient may show no
febrile response to an infection. Although there is debate
about whether or not corticosteroids increase the risk of
peptic ulcer when used alone, they act synergistically
with NSAIDs, including aspirin, to increase the risk of
serious gastrointestinal adverse effects. Latent tuberculosis
may be re-activated and patients on corticosteroids
should be advised to avoid contact with varicella zoster
virus if they are not immune.
Management of glucocorticoid withdrawal
All glucocorticoid therapy, even if inhaled or applied
topically, can suppress the HPA axis. In practice, this is
only likely to result in a crisis due to adrenal insufficiency
on withdrawal of treatment if glucocorticoids have been administered orally or systemically for longer than 3 weeks, if repeated courses have been prescribed within the previous year, or if the dose is higher than
the equivalent of 7.5 mg prednisolone per day. In these
circumstances, the drug, when it is no longer required
for the underlying condition, must be withdrawn slowly
at a rate dictated by the duration of treatment.
If glucocorticoid therapy has been prolonged, then it may
take many months for the HPA axis to recover. Allpatients must be advised to avoid sudden drug withdrawal.
They should be issued with a steroid card and/
or wear an engraved bracelet .
Recovery of the HPA axis is aided if there is no
exogenous glucocorticoid present during the nocturnal
surge in ACTH secretion.
This can be achieved by giving glucocorticoid in the morning. Giving ACTH to stimulate adrenal recovery is of no value, as the pituitary remains suppressed.
In patients who have received glucocorticoids for
longer than a few weeks, it is often valuable to confirmthat the HPA axis is recovering during glucocorticoid
withdrawal. Once the dose of glucocorticoid is reduced
to a minimum (e.g. 4 mg prednisolone or 0.5 mg dexamethasone per day), then measure plasma cortisol at
0900 hrs before the next dose. If this is detectable, then
perform an ACTH stimulation test to confirm that glucocorticoids can be withdrawn completely. Even once glucocorticoids have been successfully withdrawn, short-term replacement therapy is often advised during significant intercurrent illness occurring in subsequent months, as the HPA axis may not be able to respond fully to severe stress.
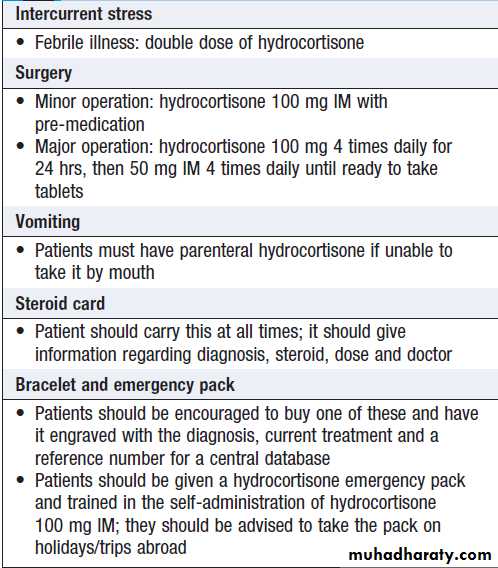
Advice to patients on glucocorticoid
replacement therapyAdrenal insufficiency
Adrenal insufficiency results from inadequate secretionof cortisol and/or aldosterone. It is potentially fatal and
notoriously variable in its presentation. A high index of
suspicion is therefore required in patients with unexplained
fatigue, hyponatraemia or hypotension. Causes
are shown in Box. The most common is ACTH
deficiency (secondary adrenocortical failure), usually
because of inappropriate withdrawal of chronic glucocorticoid therapy or a pituitary tumour . Congenital adrenal hyperplasias and Addison’s disease
(primary adrenocortical failure) are rare causes.
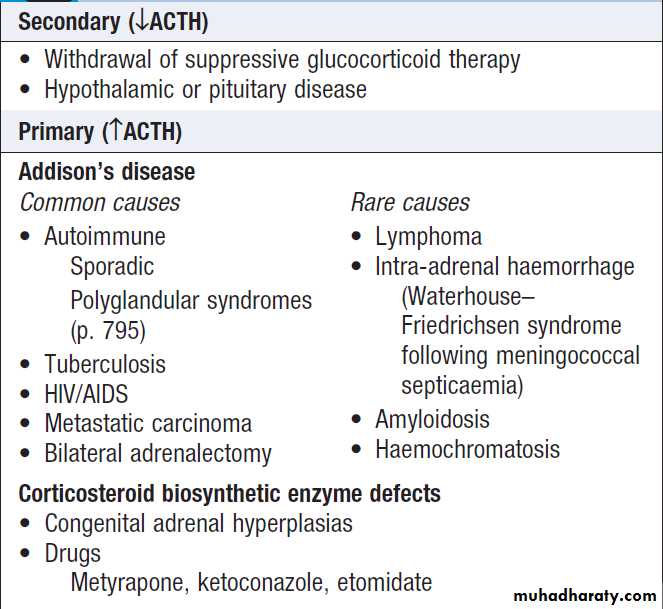
Causes of adrenocortical insufficiency
Clinical assessment
The clinical features of adrenal insufficiency are shown
in Box. In Addison’s disease, either glucocorticoid
or mineralocorticoid deficiency may come first, but
eventually all patients fail to secrete both classes of
corticosteroid.
Patients may present with chronic features and/or
in acute circulatory shock. With a chronic presentation,
initial symptoms are often misdiagnosed as chronic fatigue syndrome or depression. Adrenocortical insufficiency should also be considered in patients with hyponatraemia, even in the absence of symptoms.
Features of an acute adrenal crisis include circulatory
shock with severe hypotension, hyponatraemia, hyperkalaemia and, in some instances, hypoglycaemia and hypercalcaemia. Muscle cramps, nausea, vomiting, diarrhea and unexplained fever may be present. The crisis is often precipitated by intercurrent disease, surgery or infection.Vitiligo occurs in 10–20% of patients with autoimmune
Addison’s disease.
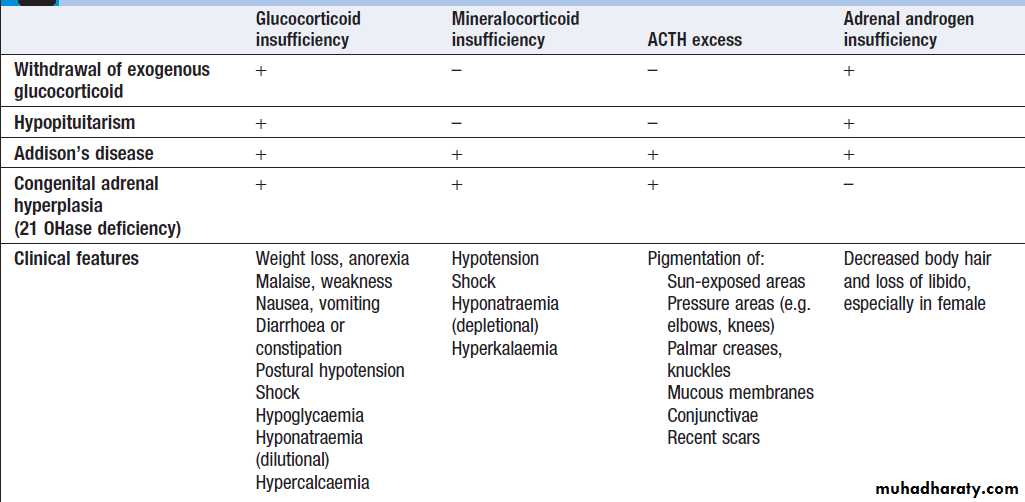
Clinical and biochemical features of adrenal insufficiency
InvestigationsTreatment should not be delayed to wait for results in
patients with suspected acute adrenal crisis. Here, a random blood sample should be stored for subsequent measurement of cortisol and, if the patient’s clinical condition permits, it may be appropriate to spend 30 minutes performing a short ACTH stimulation test before administering hydrocortisone. Investigations should be performed before treatment is given in patients who present with features suggestive of chronic adrenal insufficiency.
Assessment of glucocorticoids
Random plasma cortisol is usually low in patients with
adrenal insufficiency but it may be within the reference range, yet inappropriately low, for a seriously ill patient.
Random measurement of plasma cortisol cannot therefore
be used to confirm or refute the diagnosis, unless
the value is above 500 nmol/L (> 18 μg/ dL), which
effectively excludes adrenal insufficiency.
More useful is the short ACTH stimulation test
(also called the tetracosactrin or short Synacthen test) .
Cortisol levels fail to increase in response to exogenous ACTH in patients with primary or secondary adrenal insufficiency.
These can be distinguished by measurement of ACTH (which is low in ACTH deficiency and high in Addison’s disease).
Assessment of mineralocorticoids
Cannot be adequately assessed by electrolyte ,since hyponatraemia occurs in both aldosterone and cortisol deficiency (see Box). Hyperkalaemia is common, but not universal, in aldosterone deficiency. Plasma renin and aldosterone should be measured in the supine position. Inmineralocorticoid deficiency, plasma renin activity is
high, with plasma aldosterone being either low or in the
lower part of the reference range.
Assessment of adrenal androgens
This is not necessary in men because testosterone from
the testes is the principal androgen. In women, DHEA and androstenedione may be measured.
Other tests to establish the cause
Patients with unexplained secondary adrenocorticalinsufficiency should be investigated . In patients with elevated ACTH, further tests are required to establish the cause of Addison’s disease.
Adrenal autoantibodies are frequently positive in
autoimmune adrenal failure. If antibody tests are negative,
imaging of the adrenal glands with CT or MRI is indicated. Tuberculosis causes adrenal calcification, visible on plain X-ray or ultrasound scan. An HIV test should be performed if risk factors for infection are present . Adrenal metastases are a rare cause of adrenal insufficiency. Patients with evidence of autoimmune adrenal failure should be screened for other organ-specific autoimmune diseases, such as thyroid disease, pernicious anaemia and type 1D.
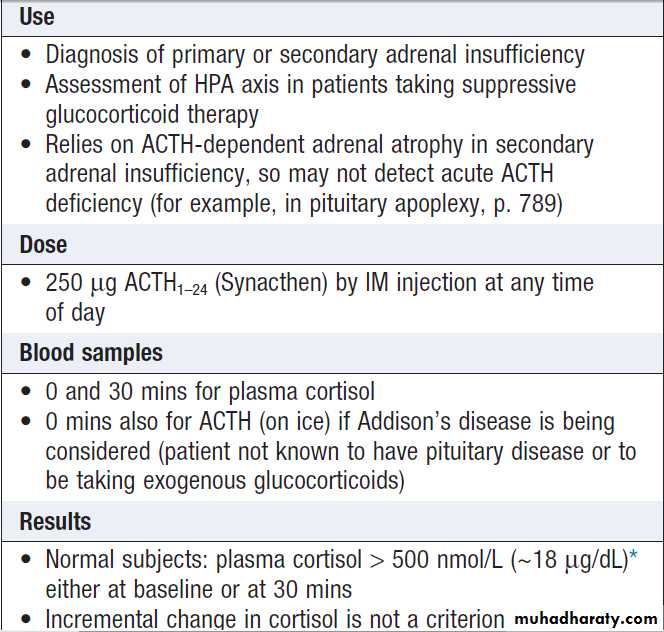
How and when to do an ACTH
stimulation test
Management
Patients with adrenocortical insufficiency always needglucocorticoid replacement therapy and usually, but not always, mineralocorticoid therapy. There is some
evidence that adrenal androgen replacement may also
be beneficial in women. Other treatments depend on
the underlying cause. The emergency management
of adrenal crisis is described in Box .
Glucocorticoid replacement
Adrenal replacement therapy consists of oral hydrocortisone(cortisol) 15–20 mg daily in divided doses, typically
10 mg on waking and 5 mg at around 1500 hrs.
These are physiological replacement doses which
should not cause Cushingoid side-effects. The dose may
need to be adjusted for the individual patient but this is
subjective. Excess weight gain usually indicates over-replacement, whilst persistent lethargy or hyperpigmentation may be due to an inadequate dose or lack of absorption. Measurement of plasma cortisol levels is not usually helpful. Advice to patients dependent on glucocorticoid replacement is given in Box .
Mineralocorticoid replacement
Fludrocortisone (9α-fluoro-hydrocortisone) is administered
at the usual dose of 0.05–0.15 mg daily, and adequacy
of replacement may be assessed by measurement
of blood pressure, plasma electrolytes and plasma renin.
Androgen replacement
Androgen replacement with DHEA (50 mg/day) is
occasionally given to women with primary adrenal
insufficiency who have symptoms of reduced libido and
fatigue, but the evidence in support of this is not robust
and treatment may be associated with side-effects such
as acne and hirsutism.
Management of adrenal crisis
Correct volume depletion• IV saline as required to normalise blood pressure and pulse
• In severe hyponatraemia (< 125 mmol/L) avoid increases
of plasma Na > 10 mmol/L/day to prevent pontine demyelination
• Fludrocortisone is not required during the acute phase of treatment
Replace glucocorticoids
• IV hydrocortisone succinate 100 mg stat, and 100 mg 4 times daily for first 12–24 hours
• Continue parenteral hydrocortisone (50–100 mg IM 4 times daily) until patient is well enough for reliable oral therapy
Correct other metabolic abnormalities
• Acute hypoglycaemia: IV 10% glucose
• Hyperkalaemia: should respond to volume replacement but occasionally requires specific therapy
Identify and treat underlying cause
• Consider acute precipitant, such as infection
• Consider adrenal or pituitary pathology
Glucocorticoids in old age
• Adrenocortical insufficiency: often insidious and difficult
to spot.
• Glucocorticoid therapy: especially hazardous in older
people, who are already relatively immunocompromised and susceptible to osteoporosis, diabetes, hypertension and other complications.
• ‘Physiological’ glucocorticoid replacement therapy:
increased risk of adrenal crisis because compliance may be
poor and there is a greater incidence of intercurrent illness.
Patient and carer education, with regular reinforcement of
the principles described in Box is crucial.
Incidental adrenal mass
It is not uncommon for a mass in the adrenal glandto be identified on a CT or MRI scan of the abdomen
that has been performed for another indication. Such
lesions are known as adrenal ‘incidentalomas’. They are
present in up to 10% of adults and the prevalence
increases with age. Eighty-five per cent of adrenal incidentalomas are non-functioning adrenal adenomas. The remainder includes functional tumours of the adrenal cortex (secreting cortisol, aldosterone or androgens), phaeochromocytomas, primary and secondary carcinomas, hamartomas and other rare disorders, including granulomatous infiltrations.
Clinical assessment and investigations
There are two key questions to be resolved: is the lesionsecreting hormones, and is it benign or malignant?
Patients with an adrenal incidentaloma are usually
asymptomatic. However, clinical signs and symptoms of
excess glucocorticoids , mineralocorticoids (see
below), catecholamines and, in women, androgens
should be sought. Investigations should include a dexamethasone suppression test, urine or plasma metanephrines and, in virilised women, measurement
of serum testosterone, DHEA and androstenedione.
Patients with hypertension should be investigated
for mineralocorticoid excess, as described below.
CT and MRI are equally effective in assessing the
malignant potential of an adrenal mass, using the following parameters:
• Size. The larger the lesion, the greater the malignant potential. Around 90% of adrenocortical carcinomas are over 4 cm in diameter, but specificity is poor since only approximately 25% of such lesions are malignant.
• Configuration. Homogeneous and smooth lesions are more likely to be benign. The presence of metastatic lesions elsewhere increases the risk of malignancy, but as many as two-thirds of adrenal incidentalomas in patients with cancer are benign.
• Presence of lipid. Adenomas are usually lipid-rich, resulting in an attenuation of below 10 Hounsfield Units (HU) on an unenhanced CT, and in signal dropout on chemical shift MRI.
• Enhancement. Benign lesions demonstrate rapid washout of contrast, whereas malignant lesions tend to retain contrast.
Histology in a sample obtained by CT-guided biopsy
is rarely indicated, and is not useful in distinguishing anadrenal adenoma from an adrenal carcinoma.
Biopsy is occasionally helpful, but should be avoided if either phaeochromocytoma or primary adrenal cancer is suspected in order to avoid precipitation of a hypertensive
crisis or seeding of tumour cells, respectively.
Management
Functional lesions and tumours of > 4 cm in diameter should be considered for removal by surgery. In patients with radiologically benign, non-functioning lesions < than 4 cm in diameter, surgery is only required if serial imaging suggests tumour growth. Optimal management of patients with low-grade cortisol secretion, as demonstrated by the dexamethasone suppression test, remains to be established.
Primary hyperaldosteronism
It may occur in as many as 10% of people with hypertension. Indications to test for mineralocorticoid excess in hypertensive patients include hypokalaemia(including hypokalaemia induced by thiazide
diuretics), poor control of blood pressure with conventional
therapy, a family history of early-onset hypertension,
or presentation at a young age. It is important to differentiate primary, caused by an intrinsic abnormality of the adrenal glands resulting in aldosterone excess, from secondary hyperaldosteronism, which is usually a consequence of enhanced activity of renin in response to inadequate renal perfusion and hypotension.
Most individuals with primary hyperaldosteronism have bilateral adrenal hyperplasia (idiopathic hyperaldosteronism), while only a minority have an aldosterone-producing adenoma (APA; Conn’s syndrome). Glucocorticoid-suppressible hyperaldosteronism
is a rare autosomal dominant condition in which aldosterone is secreted ‘ectopically’ from the adrenal fasciculata/ reticularis in response to ACTH.
Rarely, the mineralocorticoid receptor pathway in the
distal nephron is activated, even though aldosterone
concentrations are low.
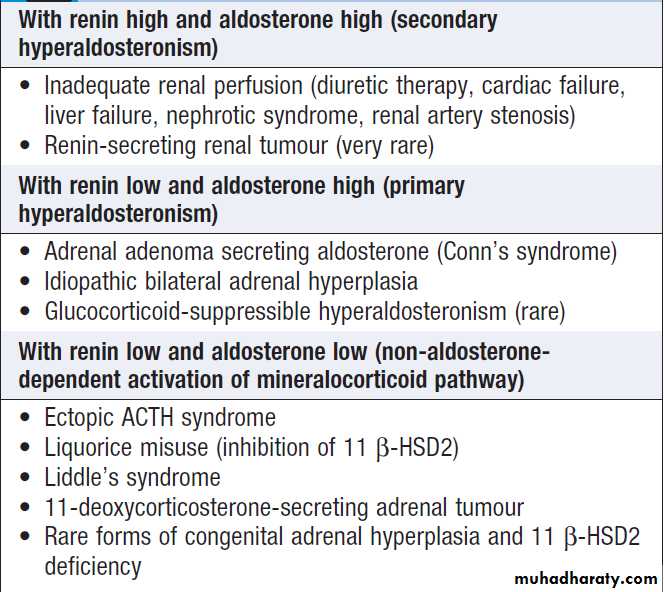
Causes of mineralocorticoid excess
Clinical featuresIndividuals with primary hyperaldosteronism are
usually asymptomatic but may have features of sodium
retention or potassium loss. Sodium retention may cause
oedema, while hypokalaemia may cause muscle weakness (or even paralysis), polyuria (secondary to renal tubular damage, which produces nephrogenic diabetes insipidus) and occasionally tetany (because of associated metabolic alkalosis and low ionised calcium).
Blood pressure is elevated but accelerated
phase hypertension is rare.
Investigations
BiochemicalRoutine blood tests may show a hypokalaemic alkalosis.
Sodium is usually at the upper end of the reference
range in primary hyperaldosteronism, but is characteristically low in secondary hyperaldosteronism (because low plasma volume stimulates antidiuretic hormone
(ADH) release and high angiotensin II levels stimulate
thirst). The key measurements are plasma renin and
aldosterone and in many centres, the aldosterone : renin ratio (ARR) is employed as a screening test for primary hyperaldosteronism in hypertensive patients. Almost all antihypertensive drugs interfere with this ratio (β-blockers inhibit whilst thiazide diuretics stimulate renin secretion).
Thus, individuals with an elevated ARR require further testing after stopping antihypertensive drugs for at least 2 weeks. If necessary, antihypertensive agents that have minimal effects on the renin–angiotensin system, such as calcium antagonists and α-blockers, may be substituted. Oral potassium supplementation may also be required, as hypokalaemia itself suppresses renin activity. If, on repeat testing, plasma renin is low and aldosterone concentrations are elevated, then further investigation under specialist
supervision may include suppression tests (sodium
loading) and/or stimulation tests (captopril or furosemide
administration) to differentiate angiotensin II-dependent aldosterone secretion in idiopathic hyperplasia from autonomous aldosterone secretion typical of an APA.
Imaging and localisation
Imaging with CT or MRI will identify most APAs , but it is important to recognise the risk of false-positives (non-functioning adrenal adenomas are common) and false-negatives (imaging may have insufficient resolution to identify adenomas with diameter of less than 0.5 cm). If the imaging is inconclusive and there is an intention to proceed with surgery on the basis of strong biochemical evidence of an APA, then adrenal vein catheterisation with measurement of aldosterone (and cortisol to confirm positioning of the catheters) is required. In some centres, this is performed even in the presence of a unilateral ‘adenoma’, to avoid inadvertent removal of an incidental non-functioning adenoma contralateral to the inapparent cause of aldosterone excess.Management
Mineralocorticoid receptor antagonists (spironolactoneand eplerenone) are valuable in treating both hypokalaemia and hypertension. Up to 20% of males develop gynaecomastia on spironolactone. Amiloride (10–40 mg/day), which blocks the epithelial sodium channel regulated by aldosterone, is an alternative.
In patients with an APA, medical therapy is usually
given for a few weeks to normalise whole-body electrolyte
balance before unilateral adrenalectomy. Laparoscopic
surgery cures the biochemical abnormality but,
depending on the pre-operative duration, hypertension
remains in as many as 70% of cases, probably because of
irreversible damage to the systemic microcirculation.
Phaeochromocytoma and paraganglioma
These are rare neuro-endocrine tumours that may secretecatecholamines (adrenaline/epinephrine, noradrenaline/
norepinephrine). Approximately 80% of these tumours
occur in the adrenal medulla (phaeochromocytomas),
while 20% arise elsewhere in the body in sympathetic
ganglia (paragangliomas). Most are benign but approximately 15% show malignant features. Around 30% are associated with inherited disorders, including neurofibromatosis,von Hippel–Lindau syndrome and MEN2 . Paragangliomas are particularly associated with mutations in the succinate dehydrogenase B, C and D genes.
Clinical features
Some patients present with hypertension, although it has been estimated that phaeochromocytoma accounts for less than 0.1% of cases of hypertension. The presentation may be with a complication of hypertension, such as stroke, myocardial infarction, left ventricular failure, hypertensive retinopathy or accelerated phase hypertension.
The apparent paradox of postural hypotension
between episodes is explained by ‘pressure natriuresis’
during hypertensive episodes so that intravascular
volume is reduced. There may also be features of the
familial syndromes associated with phaeochromocytoma.
Paragangliomas are often non-functioning.
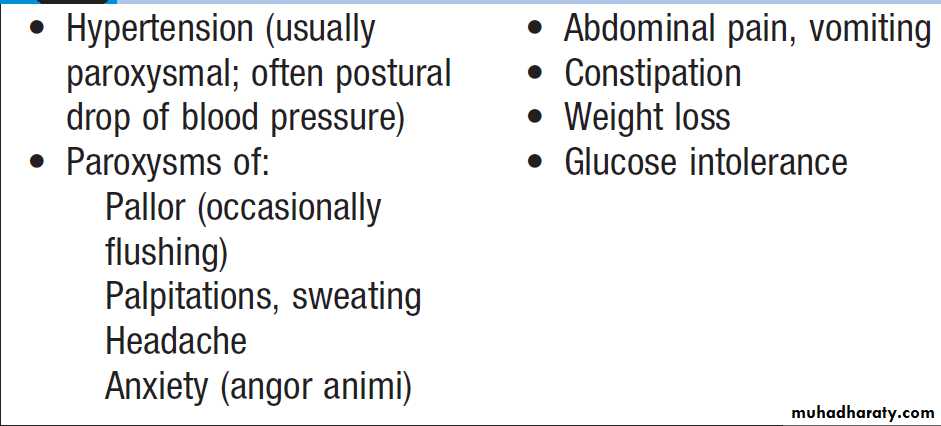
Clinical features of phaeochromocytoma
InvestigationsExcessive secretion of catecholamines can be confirmed
by measuring metabolites in plasma and/or urine (metanephrine and normetanephrine). There is a
high ‘false-positive’ rate, as misleading metanephrine
concentrations may be seen in stressed patients (during
acute illness, following vigorous exercise or severe
pain) and following ingestion of some drugs such
as tricyclic antidepressants. For this reason, a repeat
sample should usually be requested if elevated levels
are found, although, as a rule, the higher the concentration
of metanephrines, the more likely is the diagnosis
of phaeochromocytoma/paraganglioma.
Serum chromogranin A is often elevated and may be a useful tumour marker in patients with non-secretory tumours and/ or metastatic disease. Genetic testing should be considered in individuals with other features of a genetic syndrome, with a family history of phaeochromocytoma/ paraganglioma, and in those presenting < 50 years.
Localisation
Phaeochromocytomas are usually identified by abdominal
CT or MRI . Localisation of paragangliomas may be more difficult. Scintigraphy using meta- iodobenzyl guanidine (MIBG) can be useful, particularly if combined with CT. 18F-deoxyglucose PET is especially useful for detection of malignant disease and for confirming an imaging abnormality as a paraganglioma in an individual with underlying risk due to genetic mutation.
Management
Medical therapy is required to prepare the patient forsurgery, preferably for a minimum of 6 weeks to allow
restoration of normal plasma volume. The most useful
drug in the face of very high circulating catecholamines
is the α-blocker phenoxybenzamine (10–20 mg orally
3–4 times daily) because it is a non-competitive antagonist,
unlike prazosin or doxazosin. If α-blockade produces
a marked tachycardia, then a β-blocker such as
propranolol can be added. On no account should a
β-blocker be given before an α-blocker, as this may cause
a paradoxical rise in blood pressure due to unopposed
α-mediated vasoconstriction.
During surgery, sodium nitroprusside and the
short-acting α-antagonist phentolamine are useful incontrolling hypertensive episodes, which may result
from anaesthetic induction or tumour mobilisation.
Post-operative hypotension may occur and require
volume expansion and, very occasionally, noradrenaline
(norepinephrine) infusion, but is uncommon if the
patient has been prepared with phenoxybenzamine.
Metastatic tumours may behave in an aggressive or
a very indolent fashion. Management options include
debulking surgery, radionuclide therapy with 131I-MIBG,
chemotherapy and (chemo)embolisation of hepatic
metastases; some may respond to tyrosine kinase and
angiogenesis inhibitors.
Congenital adrenal hyperplasia
Pathophysiology and clinical features
Inherited defects in enzymes of the cortisol biosynthetic
pathway result in insufficiency of hormones downstream of the block, with impaired negative feedback and increased ACTH secretion. ACTH then stimulates the production of steroids upstream of the enzyme block. This produces adrenal hyperplasia and a combination of clinical features that depend on the severity and site of the defect in biosynthesis. All of these enzyme abnormalities are inherited as autosomal recessive traits.
The most common enzyme defect is 21-hydroxylase
deficiency. This results in impaired synthesis of cortisol and aldosterone and accumulation of 17OHprogesterone,which is then diverted to form adrenal androgens. In about one-third of cases, this defect is severe and presents in infancy with features of glucocorticoid and mineralocorticoid deficiency and androgen excess, such as ambiguous genitalia in girls.
In the other two-thirds, mineralocorticoid secretion is adequate but there may be features of cortisol insufficiency and/or ACTH and androgen excess, including precocious pseudo-puberty, which is distinguished from ‘true’ precocious puberty by low gonadotrophins.
Sometimes the mildest enzyme defects are not apparent until adult life, when females may present with amenorrhoea and/or hirsutism .This is
called ‘non-classical’ or ‘late-onset’ congenital adrenal
hyperplasia.
Defects of all the other enzymes in Figure are rare.
Both 17-hydroxylase and 11 β-hydroxylase
deficiency may produce hypertension due
to excess production of 11-deoxycorticosterone, which
has mineralocorticoid activity.
Investigations
Circulating 17OH-progesterone levels are raised in21-hydroxylase deficiency, but this may only be demonstrated after ACTH administration in late-onset cases.
To avoid salt-wasting crises in infancy, 17OHprogesterone
can be routinely measured in heelprick blood spot samples taken from all infants in the first week of life. Assessment is otherwise as described for adrenal insufficiency.
In siblings of affected children, antenatal genetic
diagnosis can be made by amniocentesis or chorionic
villus sampling. This allows prevention of virilisation of
affected female fetuses by administration of dexamethasone to the mother to suppress ACTH levels.
Management
The aim is to replace deficient corticosteroids and to
suppress ACTH-driven adrenal androgen production.
A careful balance is required between adequate suppression of adrenal androgen excess and excessive
glucocorticoid replacement resulting in features of
Cushing’s syndrome. In children, growth velocity is an
important measurement, since either under- or over-replacement with glucocorticoids suppresses growth. In adults, there is no uniformly agreed adrenal replacement regime, and clinical features (menstrual cycle, hirsutism, weight gain, blood pressure) and biochemical profiles (plasma renin, 17OH-progesterone and testosterone levels) provide a guide. If hirsutism is the main problem, anti-androgen therapy may be just as effective.
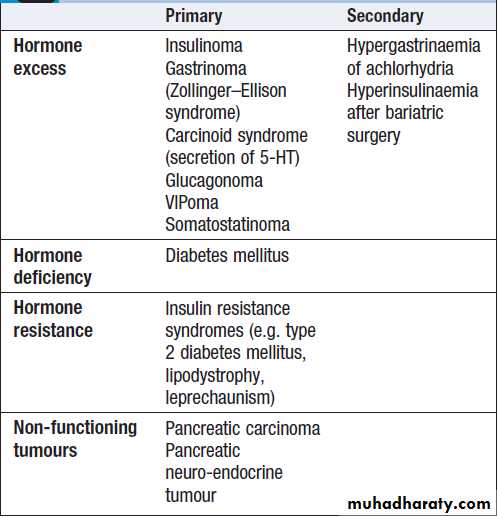
Classification of endocrine diseases of
the pancreas and gastrointestinal tractPresenting problems in endocrine pancreas disease
Spontaneous hypoglycaemiaHypoglycaemia most commonly occurs as a side-effect
of treatment with insulin or sulphonylurea drugs in people with diabetes mellitus. In non-diabetic individuals, symptomatic hypoglycaemia is rare, but it is not uncommon to detect venous blood glucose concentrations below 3.0 mmol/L in asymptomatic patients. For this reason, and because the symptoms of hypoglycaemia are non-specific, a hypoglycaemic disorder should only be diagnosed if all three conditions of Whipple’s triad are met .There is no specific blood glucose concentration at which spontaneous hypoglycaemia can be said to occur, although the lower the blood glucose concentration, the more likely it is to have
pathological significance.
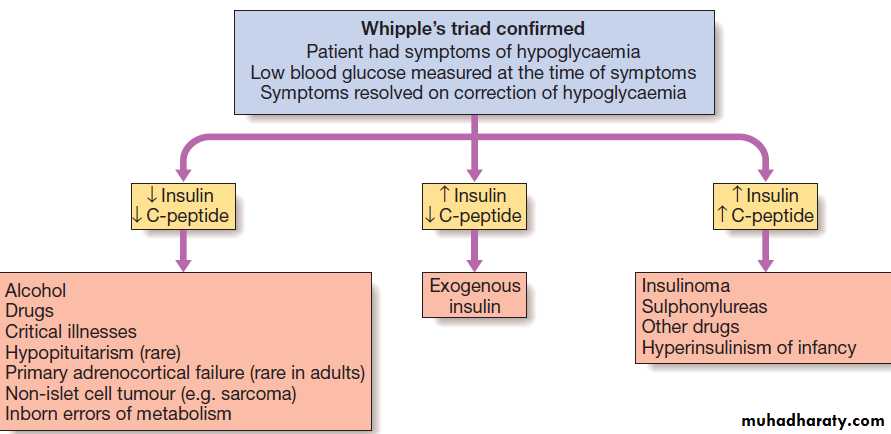
Differential diagnosis of spontaneous hypoglycaemia. Measurement of insulin and C-peptide concentrations during an episode is helpful in determining the underlying cause.
Clinical assessment
The clinical features of hypoglycaemia are described. Individuals with chronic spontaneous hypoglycaemiaoften have attenuated autonomic responses and
‘hypoglycaemia unawareness’, and may present with a
wide variety of features of neuro-glycopenia, including
odd behaviour and convulsions. The symptoms are usually episodic and relieved by consumption of carbohydrate. Symptoms occurring while fasting (such as before breakfast) or following exercise are much more likely to be representative of pathological hypoglycaemia than those which develop after food (post-prandial or ‘reactive’ symptoms). Hypoglycaemia should be considered in all comatose patients, even if there is an apparently obvious cause, such as hemiplegic stroke or alcohol intoxication.
Investigations
Does the patient have a hypoglycaemic disorder?Patients who present acutely with confusion, coma or
convulsions should be tested for hypoglycaemia at the bedside with a capillary blood sample and an automated meter. While this is sufficient to exclude hypoglycaemia,
blood glucose meters are relatively inaccurate
in the hypoglycaemic range and the diagnosis should
always be confirmed by a laboratory-based glucose
measurement. At the same time, a sample should be
taken for later measurement, if necessary, of alcohol,
insulin, C-peptide, cortisol and sulphonylurea levels. Taking these samples during an acute presentation prevents subsequent unnecessary dynamic tests and is of medico-legal importance in cases where poisoning is suspected.
Patients who attend the outpatient clinic with episodic symptoms suggestive of hypoglycaemia present a more challenging problem. The main diagnostic test is the prolonged (72-hour) fast. If symptoms of hypoglycaemia develop during the fast, then blood samples should be taken to confirm hypoglycaemia and for later measurement of insulin and C-peptide.
Hypoglycaemia is then corrected with oral or intravenous glucose and Whipple’s triad completed by confirmation of the resolution of symptoms.
The absence of clinical and biochemical evidence of hypoglycaemia during a prolonged fast effectively excludes the diagnosis of a hypoglycaemic disorder.
What is the cause of the hypoglycaemia?
In the acute setting, the underlying diagnosis is oftenobvious. In non-diabetic individuals, alcohol excess is
the most common cause of hypoglycaemia in the UK,
but other drugs – for example, salicylates, quinine and
pentamidine – may also be implicated. Hypoglycaemia
is one of many metabolic derangements which occur in
patients with hepatic failure, renal failure, sepsis or malaria. Hypoglycaemia in the absence of insulin, or any insulin-like factor, in the blood indicates impaired gluconeogenesis and/or availability of glucose from glycogen in the liver. Hypoglycaemia associated with high insulin and low C-peptide concentrations is indicative of administration of exogenous insulin, either factitiously or feloniously.
Adults with high insulin and C-peptide concentrations during an episode of hypoglycaemia are most likely to have an insulinoma, but sulphonylurea ingestion should also be considered . Usually, insulinomas in the
pancreas are small (< 5 mm diameter) but can be identified by CT, MRI or ultrasound (endoscopic or laparoscopic). Imaging should include the liver since around 10% of insulinomas are malignant. Rarely, large non-pancreatic tumours, such as sarcomas, may cause recurrent hypoglycaemia because of their ability to produce excess pro-insulin-like growth factor-2 (pro-IGF-2).
Management
Treatment of acute hypoglycaemia should be initiatedas soon as laboratory blood samples have been taken,
and should not be deferred until formal laboratory confirmation has been obtained.
Intravenous dextrose (5% or 10%) is effective in the short term in the obtunded patient, and should be followed on recovery with oral unrefined carbohydrate (starch).
Continuous dextrose infusion may be necessary, especially in sulphonylurea poisoning.
IM glucagon (1 mg) stimulates hepatic glucose release, but is ineffective in patients with depleted glycogen reserves, such as in alcohol excess or liver disease.
Chronic recurrent hypoglycaemia in insulin-secreting
tumours can be treated by regular consumption of oral
carbohydrate combined with agents that inhibit insulin
secretion (diazoxide or somatostatin analogues).
Insulinomas are resected when benign, providing the individual is fit enough to undergo surgery.
Metastatic malignant insulinomas are incurable and are managed along the same lines as other metastatic neuro-endocrine tumours (see below).

Spontaneous hypoglycaemia in old age
Gastroenteropancreatic neuro-endocrine tumoursNeuro-endocrine tumours (NETs) are a heterogeneous
group derived from neuro-endocrine cells in many
organs, including the gastrointestinal tract, lung,
adrenals (phaeochromocytoma) and thyroid
(medullary carcinoma). Most NETs occur sporadically, but a proportion are associated with genetic cancer syndromes, such as MEN 1, MEN 2 and neurofibromatosis type 1.
NETs may secrete hormones into the circulation. Gastroenteropancreatic NETs arise in organs that are derived embryologically from the gastrointestinal tract.
The term ‘carcinoid’ is often used when referring to non-pancreatic gastroenteropancreatic NETs because, when initially described, they were thought to behave in an indolent fashion compared with conventional cancers. It is now recognised that there is a wide spectrum of malignant potential for all NETs; some are benign (most insulinomas and appendiceal carcinoid tumours), while others have an aggressive clinical course with widespread metastases (small-cell carcinoma of the lung).
The majority behave in an intermediate manner, with relatively slow growth but a propensity to invade and metastasise to remote organs, especially the liver.
Clinical features
Patients with gastroenteropancreatic NETs often have a
history of abdominal pain over many years prior to diagnosis and usually present with local mass effects, such
as small-bowel obstruction, appendicitis, and pain from
hepatic metastases. Thymic and bronchial carcinoids
occasionally present with ectopic ACTH syndrome.
Pancreatic NETs can also cause hormone excess but most are non-functional. The classic ‘carcinoid syndrome’ occurs when vasoactive hormones reach the systemic circulation. In gastrointestinal carcinoids, this invariably means that the tumour has metastasised to the liver or peritoneum, which allow secreted hormones to access the systemic circulation; hormones secreted metabolised and inactivated in the liver.
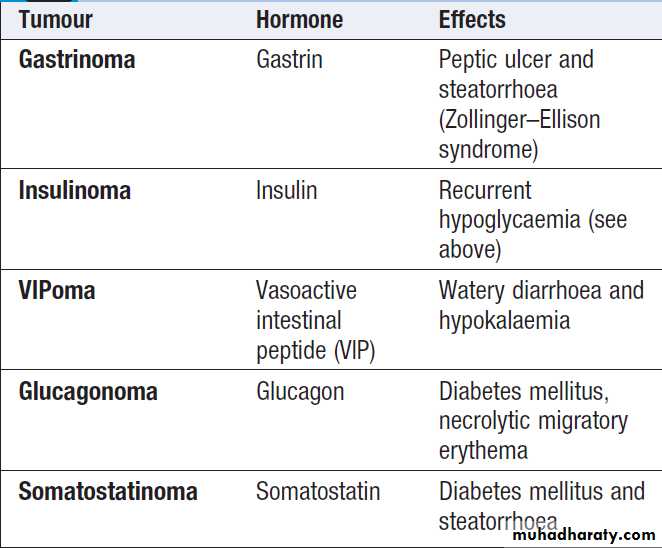
Pancreatic neuro-endocrine tumours

Clinical features of the carcinoid syndrome
InvestigationsA combination of imaging with ultrasound, CT, MRI
and/or radio-labelled somatostatin analogue will usually identify the primary tumour and allow staging, which is crucial for determining prognosis.
Biopsy of the primary tumour or a metastatic deposit
is required to confirm the histological type. NETs
demonstrate immunohistochemical staining for the
proteins chromogranin A and synaptophysin, and
the histological grade may also provide prognostic
information.
Carcinoid syndrome is confirmed by measuring elevated
concentrations of 5-hydroxyindoleacetic acid
(5-HIAA), a metabolite of serotonin, in a 24-hour urine
collection. False-positives can occur, particularly if the
individual has been eating certain foods, such as avocado
and pineapple.
Plasma chromogranin A can be measured in a fasting blood sample, along with the hormones listed in Box.
All of these can be useful as tumour markers.
Management
Treatment of solitary tumours is by surgical resection. Ifmetastatic or multifocal primary disease is present,
then surgery is usually not indicated, unless there is a
complication such as gastrointestinal obstruction. Diazoxide
can reduce insulin secretion in insulinomas, and
high doses of proton pump inhibitors suppress acid production in gastrinomas. Somatostatin analogues are
effective in reducing the symptoms of carcinoid syndrome
and of excess glucagon and vasoactive intestinal
peptide (VIP) production. The slow-growing nature of
NETs means that conventional cancer therapies, such as
chemotherapy and radiotherapy, have limited efficacy.
Other treatments, such as interferon, targeted radionuclide therapy with 131I-MIBG and radio-labelled
somatostatin analogues (which may be taken up by
NET metastases), and resection/embolisation of hepatic
metastases, may have a role in the palliation of symptoms
but there is little evidence that they prolong life.
The tyrosine kinase inhibitor sunitinib and the mammalian
target of rapamycin (mTOR) inhibitor everolimus have shown benefit in progressive pancreatic NETS and should be considered as part of standard therapy.

Tyrosine kinase/mTOR inhibitors in advanced pancreatic neuro-endocrine tumours
THE HYPOTHALAMUS AND THE PITUITARY GLANDDiseases of the hypothalamus and pituitary have
an annual incidence of approximately 3 : 100 000 and a prevalence of 30–40 per 100 000. The pituitary plays a
central role in several major endocrine axes, so that
investigation and treatment invariably involve several
other endocrine glands.
Functional anatomy, physiology and investigations
The pituitary gland is enclosed in the sella turcica and bridged over by a fold of dura mater called the diaphragma sellae, with the sphenoidal air sinuses below and the optic chiasm above. The cavernous sinuses are lateral to the pituitary fossa and contain the 3rd, 4th and 6th cranial nerves and the internal carotid arteries.The gland is composed of, anterior and posterior lobes, connected to the hypothalamus by the infundibular stalk, which has portal vessels carrying blood from the median eminence of the hypothalamus to the anterior lobe and nerve fibres to the posterior lobe. By far the most common disorder is an adenoma of the anterior pituitary gland.
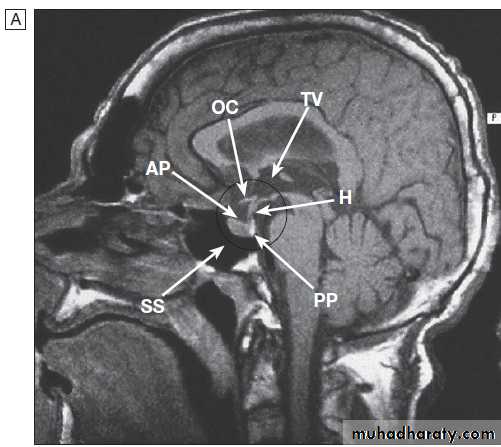
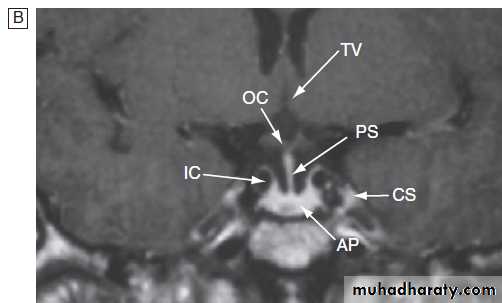
Anatomical relationships of the normal pituitary gland and hypothalamus.
A Sagittal MRI.B Coronal MRI. (AP = anterior pituitary; CS = cavernous sinus; H =
hypothalamus; IC = internal carotid artery; OC = optic chiasm; PP =
posterior pituitary; PS = pituitary stalk; SS = sphenoid sinus; TV = third ventricle)
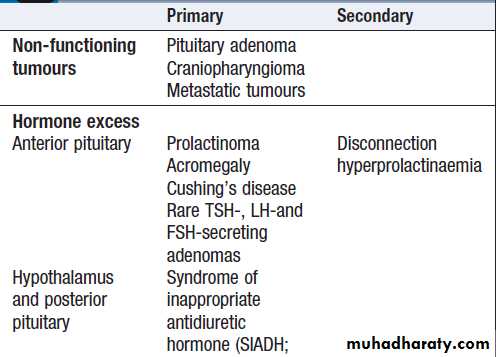
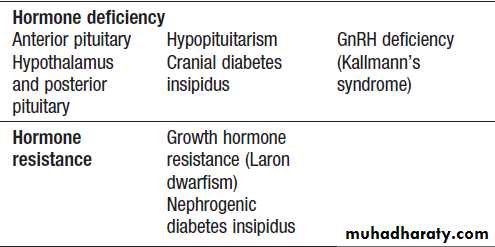
Classification of diseases of the pituitary
and hypothalamusInvestigation of patients with pituitary disease
The approach to investigation is similar in all cases . Tests for hormone excess vary according to the hormone in question. For example, prolactin is not secreted in pulsatile fashion, although it rises with significant psychological stress.Assuming that the patient was not distressed by
venepuncture, a random measurement of serum prolactin
is sufficient to diagnose hyperprolactinaemia. In contrast,
growth hormone is secreted in a pulsatile fashion. A high random level does not confirm acromegaly;
the diagnosis is only confirmed by failure of growth
hormone to be suppressed during an oral glucose tolerance
test, and a high serum insulin-like growth factor-1(IGF-1).
Similarly, in suspected ACTH-dependent Cushing’s
disease , random plasma cortisol is unreliable and the diagnosis is usually made by a dexamethasone suppression test. The most common local complication of a large pituitary tumour is compression of the optic pathway. The
resulting visual field defect can be documented using a
Goldman’s perimetry chart.
MRI reveals ‘abnormalities’ of the pituitary gland
in as many as 10% of ‘healthy’ middle-aged people.
It should therefore be performed only if there is a
clear biochemical abnormality or in a patient who
presents with clinical features of pituitary tumour.
A pituitary tumour may be classified as either a
macroadenoma (> 10 mm diameter) ora microadenoma (< 10 mm diameter).
Surgical biopsy is usually only performed as part of
a therapeutic operation. Conventional histology identifies
tumours as chromophobe (usually non-functioning),
acidophil (typically prolactin- or growth hormone-secreting)
or basophil (typically ACTH-secreting); immunohistochemistry may confirm their secretory
capacity but is poorly predictive of growth potential.
Identify pituitary hormone deficiency
ACTH deficiency• Short ACTH stimulation test
• Insulin tolerance test only if uncertainty in interpretation of short ACTH stimulation test
(e.g. acute presentation)
LH/FSH deficiency
• In the male, measure random serum testosterone, LH and FSH
• In the pre-menopausal female, ask if menses are regular
• In the post-menopausal female, measure random serum LH and FSH (which would normally be > 30 mU/L)
How to investigate patients with suspected
pituitary hypothalamic disease
TSH deficiency
• Measure random serum T4
• Note that TSH is often detectable in secondary hypothyroidism
Growth hormone deficiency
Only investigate if growth hormone replacement therapy is being
Contemplated
• Measure immediately after exercise
• Consider other stimulatory tests
Cranial diabetes insipidus
Only investigate if patient complains of polyuria/polydipsia , which may be masked by ACTH or TSH deficiency
• Exclude other causes of polyuria with blood glucose,
potassium and calcium measurements
• Water deprivation or 5% saline infusion test
Identify hormone excess
• Measure random serum prolactin
• Investigate for acromegaly (glucose tolerance test) or
Cushing’s syndrome if there are clinical features
Establish the anatomy and diagnosis
• Consider visual field testing
• Image the pituitary and hypothalamus by MRI or CT
How to investigate patients with suspected
pituitary hypothalamic disease – cont’d
Presenting problems in hypothalamic and pituitary disease
The clinical features of pituitary disease are shown inFigure overleaf. Younger women with pituitary
disease most commonly present with secondary amenorrhoea or galactorrhoea (in hyperprolactinaemia).
Post-menopausal women and men of any age
are less likely to report symptoms of hypogonadism and
so are more likely to present late with larger tumours
causing visual field defects.
Nowadays, many patients present with the incidental finding of a pituitary tumour on a CT or MRI scan.
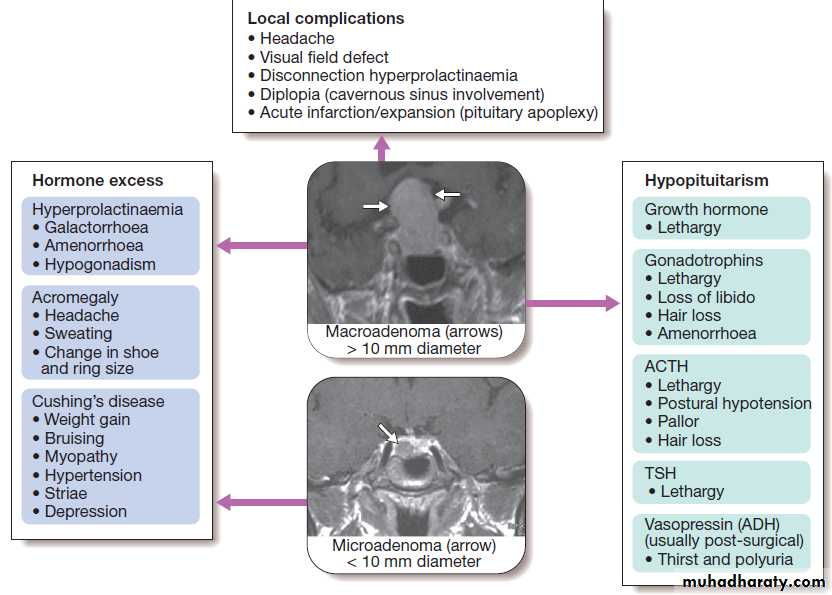
Common symptoms and signs to consider in a patient with suspected pituitary disease.
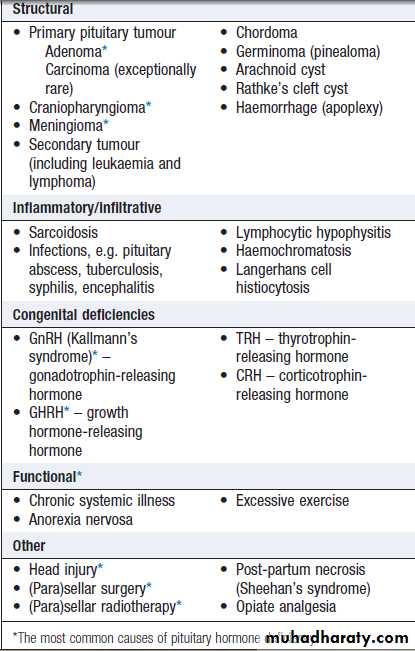
Causes of anterior pituitary hormone
deficiencyHypopituitarism
Hypopituitarism describes combined deficiency of anyof the anterior pituitary hormones.
The clinical presentation is variable and depends on the underlying lesion and the pattern of resulting hormone deficiency.
The most common cause is a pituitary macroadenoma but
other causes are listed in Box.
Clinical assessment
The presentation is highly variable. For example, followingradiotherapy to the pituitary region, there is a characteristic sequence of loss of pituitary hormone secretion.
Growth hormone secretion is often the earliest to be lost.
In adults, this produces lethargy, muscle weakness and
increased fat mass.
Next, gonadotrophin (LH and FSH) secretion becomes impaired, in the male, loss of libido and, in the female, oligomenorrhoea or amenorrhoea. Later, in the male there may be gynaecomastia and decreased frequency of shaving. In both sexes, axillary and pubic hair eventually become sparse or even absent and the skin becomes characteristically finer and wrinkled.
Chronic anaemia may also occur. The next hormone to
be lost is usually ACTH, resulting in symptoms of cortisol
insufficiency (postural hypotension and a dilutional hyponatraemia). In contrast to primary adrenal insufficiency , angiotensin II-dependent zona glomerulosa function is not lost and hence aldosterone secretion maintains normal plasma potassium. In contrast to the pigmentation of Addison’s disease due to high levels of circulating ACTH acting on the skin melanocytes, a striking degree of pallor is usually present.
Finally, TSH secretion is lost with consequent secondary hypothyroidism.
This contributes further to apathy and cold intolerance.
In contrast to primary hypothyroidism, frank myxoedema is rare, presumably because the thyroid retains some autonomous function.
The onset of all of the above symptoms is notoriously
insidious. However, patients sometimes present acutely
unwell with glucocorticoid deficiency.
This may be precipitated by a mild infection or injury, or may occur secondary to pituitary apoplexy .
Other features of pituitary disease may be present (Fig.)
Investigations
The strategy for investigation of pituitary disease isdescribed in Box. In acutely unwell patients, the
priority is to diagnose and treat cortisol deficiency.
Other tests can be undertaken later. Specific
dynamic tests for diagnosing hormone deficiency are
described in Boxes. More specialised biochemical tests, such as insulin tolerance tests , GnRH and TRH tests, are rarely required.
All patients with biochemical evidence of pituitary
hormone deficiency should have an MRI or CT scan to
identify pituitary or hypothalamic tumours. If a tumour is not identified, then further investigations are indicated
to exclude infectious or infiltrative causes.

Tests of growth hormone secretion
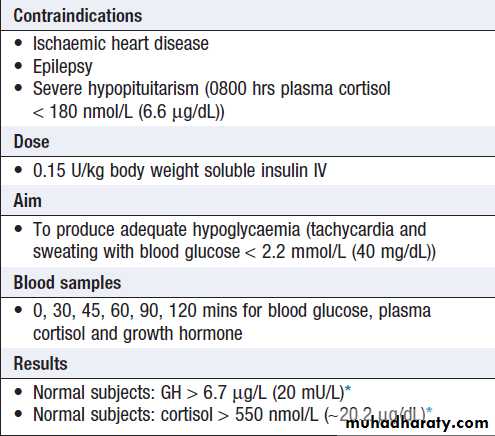


How and when to do an insulin
tolerance testManagement
Treatment of acutely ill patients is similar to thatdescribed for adrenocortical insufficiency, except that sodium depletion is not an important component to correct. Chronic hormone replacement therapies are described below. Once the cause of hypopituitarism is established, specific treatment – of a pituitary macroadenoma, for example (see below) – may be required.
Cortisol replacement
Hydrocortisone should be given if there is ACTH deficiency.
Mineralocorticoid replacement is not required.
Thyroid hormone replacement
Levothyroxine 50–150 μg once daily should be given .
Unlike in primary hypothyroidism, measuring TSH is not helpful in adjusting the replacement dose because patients with hypopituitarism often secrete glycoproteins which are
measured in the TSH assays but are not bioactive. The
aim is to maintain serum T4 in the upper part of the reference range.
It is dangerous to give thyroid replacement in adrenal insufficiency without first giving glucocorticoid therapy, since this may precipitate adrenal crisis.
Sex hormone replacement
This is indicated if there is gonadotrophin deficiency in
women under the age of 50 and in men to restore normal
sexual function and to prevent osteoporosis .
Growth hormone replacement
GH is administered by daily subcutaneous self-injection to children and adolescents with GH deficiency and, until recently, was discontinued once the epiphyses had fused. However, although hypopituitary adults receiving ‘full’ replacement with hydrocortisone, levothyroxine and sex steroids are usually much improved by these therapies, some individuals remain lethargic and unwell. Some of these patients feel better, and have objective improvements in their fat : muscle mass ratios and other metabolic parameters, if they are also given GH replacement.Treatment with GH may also help young adults to achieve a higher peak bone mineral density. The principal side-effect is sodium retention, manifest as peripheral oedema or carpal tunnel syndrome.
For this reason, GH replacement should be started at a low dose, with monitoring of the response
by measurement of serum IGF-1.
Pituitary tumour
Pituitary tumours produce a variety of mass effects,depending on their size and location, but also present
as incidental findings on CT or MRI, or with hypopituitarism, as described above. A wide variety of disorders can present as mass lesions in or around the pituitary gland .
Most intrasellar tumours are pituitary macroadenomas (most commonly nonfunctioning adenomas, see Fig.),
whereas suprasellar masses may be craniopharyngiomas (see Fig.). The most common cause of a parasellar mass is a meningioma.
Clinical assessment
Clinical features are shown in Figure. A common
but non-specific presentation is with headache, which
may be the consequence of stretching of the diaphragma
sellae. Although the classical abnormalities associated
with compression of the optic chiasm are bitemporal
hemianopia (see Fig) or upper quadrantanopia, any type of visual field defect can result from suprasellar
extension of a tumour because it may compress the optic
nerve (unilateral loss of acuity or scotoma) or the optic
tract (homonymous hemianopia).
Optic atrophy may be apparent on ophthalmoscopy.
Lateral extension of a sellar mass into the cavernous sinus with subsequent compression of the 3rd, 4th or 6th cranial nerve may cause diplopia and strabismus, but in anterior pituitary tumours this is an unusual presentation.
Occasionally, pituitary tumours infarct or there is
bleeding into cystic lesions. This is termed ‘pituitary
apoplexy’ and may result in sudden expansion with
local compression symptoms and acute-onset hypopituitarism.
Non-haemorrhagic infarction can also occur in a normal pituitary gland; predisposing factors include catastrophic obstetric haemorrhage (Sheehan’s syndrome), diabetes mellitus and raised intracranial pressure.
Investigations
Patients suspected of having a pituitary tumour shouldundergo MRI or CT. Whilst some lesions have distinctive
neuro-radiological features, the definitive diagnosis
is made on histology after surgery. All patients
with (para)sellar space-occupying lesions should have
pituitary function assessed as described in Box.
Management
Modalities of treatment of common pituitary and
hypothalamic tumours are shown in Box . Associated
hypopituitarism should be treated as described above.
Urgent treatment is required if there is evidence of pressure on visual pathways. The chances of recovery of a visual field defect are proportional to the duration of symptoms, with full recovery unlikely if the defect has been present for longer than 4 months. In the presence of a sellar mass lesion, it is crucial that serum prolactin is measured before emergency surgery is performed. If the prolactin is over 5000 mU/L, then the lesion is likely to be a macroprolactinoma and to respond to a dopamine agonist with shrinkage of the lesion, making surgery unnecessary
Most operations on the pituitary are performed using
the trans- sphenoidal approach via the nostrils, whiletransfrontal surgery via a craniotomy is reserved for
suprasellar tumours. It is uncommon to be able to resect
lateral extensions into the cavernous sinuses. All operations
on the pituitary carry a risk of damaging normal
endocrine function; this risk increases with the size of
the primary lesion.
Pituitary function should be retested 4–6 weeks following surgery, primarily to detect the development of any new hormone deficits.
Rarely, the surgical treatment of a sellar lesion can result
in recovery of hormone secretion that was deficient
pre-operatively.
Following surgery, usually after 3–6 months, imaging
should be repeated and, if there is a significant residualmass and the histology confirms an anterior pituitary
tumour, external radiotherapy may be given to reduce
the risk of recurrence. Radiotherapy is not useful in
patients requiring urgent therapy because it takes many
months or years to be effective and there is a risk of acute
swelling of the mass. Fractionated radiotherapy carries
a life-long risk of hypopituitarism (50–70% in the first
10 years) and annual pituitary function tests are obligatory.
There is also concern that radiotherapy might impair cognitive function, cause vascular changes and even induce primary brain tumours, but these side effects have not been quantified reliably and are likely to be rare.
Stereotactic radiosurgery, best delivered by the ‘gamma knife’, allows specific targeting of residual disease in a more focused fashion.
Non-functioning tumours should be followed up
by repeated imaging at intervals that depend on the size
of the lesion and on whether or not radiotherapy has
been administered. For smaller lesions that are not
causing mass effects, therapeutic surgery may not be
indicated and the lesion may simply be monitored by
serial neuroimaging without a clear-cut diagnosis
having been established.
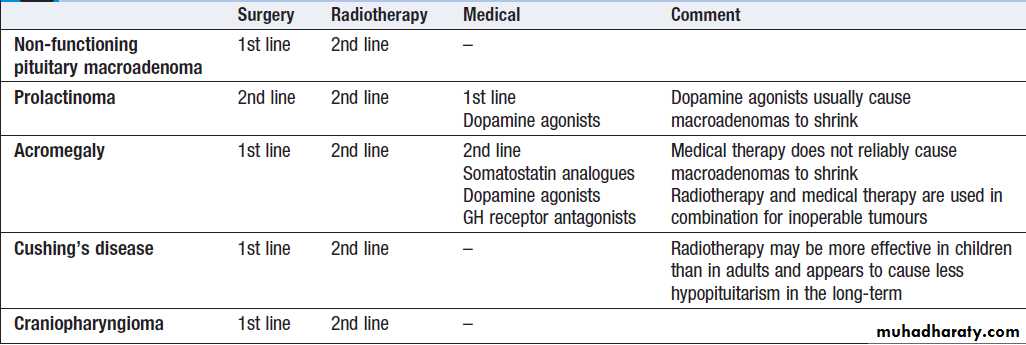
Therapeutic modalities for hypothalamic and pituitary tumours
Hyperprolactinaemia/galactorrhoeaHyperprolactinaemia is a common abnormality which
usually presents with hypogonadism and/or galactorrhoea
(lactation in the absence of breastfeeding). Since prolactin stimulates milk secretion but not breast development, galactorrhoea rarely occurs in men and only does so if gynaecomastia has been induced by hypogonadism.
The differential diagnosis is shown in Box . Many drugs, especially dopamine antagonists, elevate prolactin concentrations. Pituitary tumours can cause it by directly secreting prolactin (prolactinomas, see below), or by compressing the infundibular stalk and thus interrupting the tonic inhibitory effect of hypothalamic dopamine on prolactin secretion (‘disconnection’ hyperprolactinaemia).
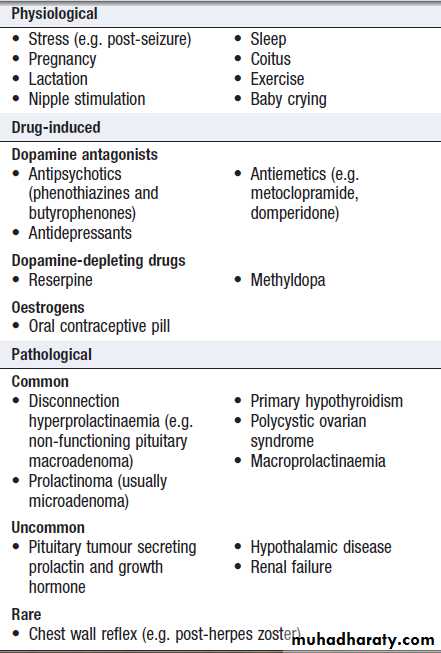
Causes of hyperprolactinaemia
Prolactin usually circulates as a free hormone in plasma, but in some becomes bound to an IgG antibody. This complex is known as macroprolactin and such patientshave macroprolactinaemia (not to be confused with macro-prolactinoma, a prolactin-secreting pituitary tumour of more than 1 cm in diameter). Since macroprolactin cannot cross blood-vessel walls to reach prolactin receptors in target tissues, it is of no pathological significance.
Identification of macroprolactin requires gel filtration chromatography or polyethylene glycol precipitation techniques, and one of these tests should be performed in all patients with hyperprolactinaemia if
the prolactin assay is known to cross-react.
Clinical assessment
In women, in addition to galactorrhoea, hypogonadismassociated with hyperprolactinaemia causes secondary
amenorrhoea and anovulation with infertility .
Important points in the history include drug use, recent
pregnancy and menstrual history. The quantity of milk
produced is variable, and it may be observed only by
manual expression. In men there is decreased libido,
reduced shaving frequency and lethargy . Unilateral
galactorrhoea may be confused with nipple discharge,
and breast examination to exclude malignancy
or fibrocystic disease is important. Further assessment
should address the features in Figure .
Investigations
Pregnancy should first be excluded before further investigations are performed in women of child-bearing
potential. The upper limit of normal for many assays of
serum prolactin is approximately 500 mU/L (14 ng/mL).
In non-pregnant and non-lactating patients, monomeric
prolactin concentrations of 500–1000 mU/L are likely to
be induced by stress or drugs, and a repeat measurement
is indicated. Levels between 1000 and 5000 mU/L
are likely to be due to either drugs, or a microprolactinoma
or ‘disconnection’ hyperprolactinaemia.
Levels above 5000 mU/L are highly suggestive of a
macroprolactinoma.Patients with prolactin excess should have tests of
gonadal function and T4 and TSH should be
measured to exclude primary hypothyroidism causing
TRH-induced prolactin excess. Unless the prolactin
falls after withdrawal of relevant drug therapy, a serum
prolactin consistently above the reference range is an
indication for MRI or CT scan of the hypothalamus and
pituitary. Patients with a macroadenoma also need tests
for hypopituitarism .
Management
If possible, the underlying cause should be corrected (for
example, cessation of offending drugs and giving levothyroxine replacement in primary hypothyroidism). If
dopamine antagonists are the cause, then dopamine
agonist therapy is contraindicated, and if gonadal dysfunction is the primary concern, sex steroid replacement
therapy may be indicated. Troublesome physiological
galactorrhoea can also be treated with dopamine agonists.
Management of prolactinomas is described below.
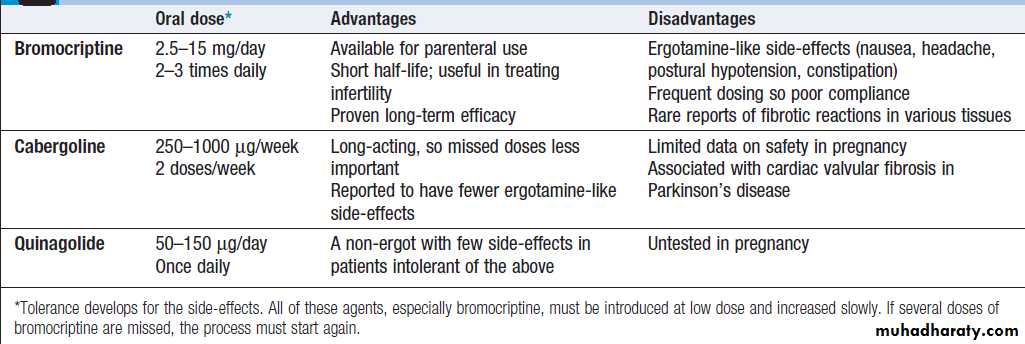
Dopamine agonist therapy: drugs used to treat prolactinomas
ProlactinomaMost prolactinomas in pre-menopausal women are
microadenomas because the symptoms of prolactin
excess usually result in early presentation. Prolactin- secreting cells of the anterior pituitary share a common
lineage with GH-secreting cells, so occasionally prolactinomas can secrete excess GH and cause acromegaly. In prolactinomas there is a relationship between prolactin concentration and tumour size: the higher the level, the bigger the tumour.
Some macroprolactinomas can elevate prolactin concentrations above 100 000 mU/L.
The investigation of prolactinomas is the same as for
other pituitary tumours (see above).
Management
As shown in Box, several therapeutic modalities
can be employed in the management of prolactinomas.
Medical
Dopamine agonist drugs are first-line therapy for the
majority of patients (Box). They usually reduce serum prolactin concentrations and cause significant tumour shrinkage after several months of therapy (Fig.) , but visual field defects, if present, may improve within days of first administration.
It is possible to withdraw dopamine agonist therapy without recurrence of hyperprolactinaemia after a few years of treatment in some patients with a microadenoma.
Also, after the menopause, suppression of prolactin is only required in microadenomas if galactorrhoea is troublesome, since hypogonadism is then physiological and tumour growth unlikely. In patients with macroadenomas, drugs can only be withdrawn after curative surgery or radiotherapy and under close supervision.
Ergot-derived dopamine agonists (bromocriptine and cabergoline) can bind to 5-HT2B receptors in the heart and elsewhere and have been associated with fibrotic reactions, particularly tricuspid valve regurgitation, when used in high doses in patients with Parkinson’s disease.
If dopamine agonist therapy is prolonged, periodic screening by echocardiography or use of non-ergot agents (quinagolide) may be indicated.
Surgery and radiotherapy
Surgical decompression is usually only necessary whena macroprolactinoma has failed to shrink sufficiently
with dopamine agonist therapy, and this may be because
the tumour has a significant cystic component. Surgery
may also be performed in patients who are intolerant of
dopamine agonists. Microadenomas can be removed selectively by trans-sphenoidal surgery with a cure rate
of about 80% but recurrence is not unusual; the cure rate
for surgery in macroadenomas is substantially lower. External irradiation may be required for macroadenomas to prevent regrowth if dopamine agonists are stopped.
Pregnancy
Hyperprolactinaemia often presents with infertility, so
dopamine agonist may be followed by pregnancy.
Patients with microadenomas should be advised to withdraw dopamine agonist therapy as soon as pregnancy is confirmed. In contrast, macroprolactinomas may enlarge rapidly under oestrogen stimulation and these patients should continue dopamine agonist therapy and need measurement of prolactin levels and visual fields during pregnancy. All patients should be advised to report headache or visual disturbance promptly.
Acromegaly
Acromegaly is caused by growth hormone (GH) secretionfrom a pituitary tumour, usually a macroadenoma,
and carries an approximate two-fold excess mortality
when untreated.
Clinical features
If GH hypersecretion occurs before puberty, then the
presentation is with gigantism. More commonly, GH
excess occurs in adult life and presents with acromegaly.
If hypersecretion starts in adolescence and persists into
adult life, then the two conditions may be combined. The
clinical features are shown in Figure. The most common complaints are headache and sweating. Additional
features include those of any pituitary tumour.
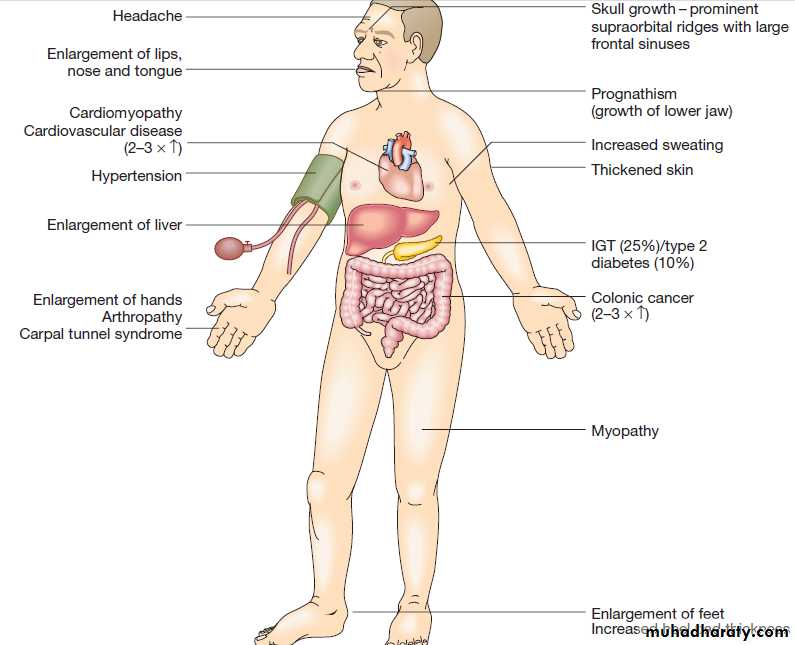
Clinical features of acromegaly. (IGT = impaired glucose tolerance
Investigations
The clinical diagnosis must be confirmed by measuringGH levels during an oral glucose tolerance test and
measuring serum IGF-1. In normal subjects, plasma GH
suppresses to below 0.5 μg/L (approximately 2 mU/L).
In acromegaly, GH does not suppress and in about 30%
of patients there is a paradoxical rise; IGF-1 is also elevated.
The rest of pituitary function should be investigated
as described in Box. Prolactin concentrations are elevated in about 30% of patients due to co-secretion of prolactin from the tumour. Additional tests in acromegaly may include screening for colonic neoplasms with colonoscopy.
Management
The main aims are to improve symptoms and to normaliseserum GH and IGF-1 to reduce morbidity and mortality.
Treatment is summarised in Box.
Surgical
Trans- sphenoidal surgery is usually the first line of treatment and may result in cure of GH excess, especially in
patients with microadenomas. More often, surgery
serves to debulk the tumour and further second-line
therapy is required, according to post-operative imaging
and glucose tolerance test results.
Radiotherapy
External radiotherapy is usually employed as second- line
treatment if acromegaly persists after surgery, to
stop tumour growth and lower GH levels. However, GH
levels fall slowly (over many years) and there is a risk
of hypopituitarism.
Medical
If acromegaly persists after surgery, medical therapy
is usually employed to lower GH levels to below 1.5 μg/L (below approximately 5 mU/L) and to normalise IGF-1 concentrations. Medical therapy may be discontinued after several years in patients who have received radiotherapy. Somatostatin analogues (such as octreotide or lanreotide) can be administered as slow-release injections every few weeks.
Somatostatin analogues can also be used as primary therapy for acromegaly either as an alternative or in advance of surgery, given evidence that they can induce modest tumour shrinkage in some patients. Dopamine agonists are less effective at lowering GH but may sometimes be helpful, especially with associated prolactin excess.
Pegvisomant is a peptide GH receptor antagonist administered by daily self-injection and may be indicated in some patients whose GH and IGF-1 concentrations fail
to suppress sufficiently following somatostatin analogue therapy.
Craniopharyngioma
Craniopharyngiomas are benign tumours that developin cell rests of Rathke’s pouch, and may be located
within the sella turcica, or commonly in the suprasellar
space. They are often cystic, with a solid component that
may or may not be calcified .In young people,
they are diagnosed more commonly than pituitary adenomas.
They may present with pressure effects on adjacent
structures, hypopituitarism and/or cranial diabetes
insipidus. Other clinical features directly related to
hypothalamic damage may also occur. These include
hyperphagia and obesity, loss of the sensation of thirst
and disturbance of temperature regulation.
Craniopharyngiomas can be treated by the trans-sphenoidal route but surgery may also involve a craniotomy, with a relatively high risk of hypothalamic
damage and other complications. If the tumour has a
large cystic component, it may be safer to place in the
cyst cavity a drain which is attached to a subcutaneous
access device, rather than attempt a resection. Whatever
form it takes, surgery is unlikely to be curative and
radiotherapy is usually given to reduce the risk of
relapse. Unfortunately, craniopharyngiomas often recur,
requiring repeated surgery. They often cause considerable morbidity, usually from hypothalamic obesity, water balance problems and/or visual failure.
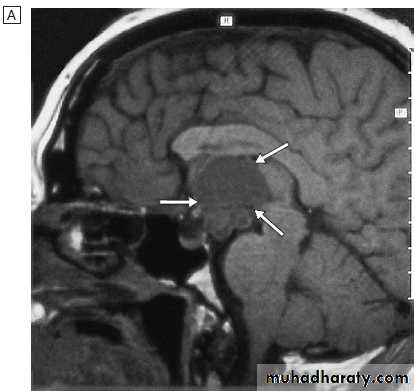
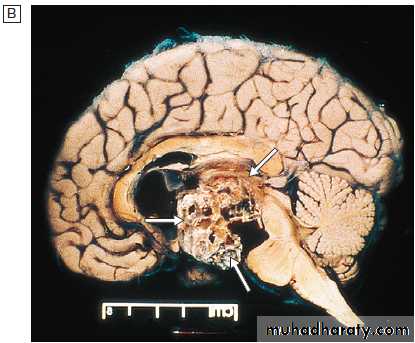
Craniopharyngioma.
A This developmental tumour characteristically presents in younger patients; it is often cystic and calcified, as shown in this MRI scan (arrows).B Pathology specimen.
Diabetes insipidus
This uncommon disorder is characterised by the persistentexcretion of excessive quantities of dilute urine
and by thirst. It is classified into two types:
• cranial diabetes insipidus, in which there is
deficient production of ADH by the hypothalamus
• nephrogenic diabetes insipidus, in which the renal
tubules are unresponsive to ADH.
The underlying causes are listed in Box.
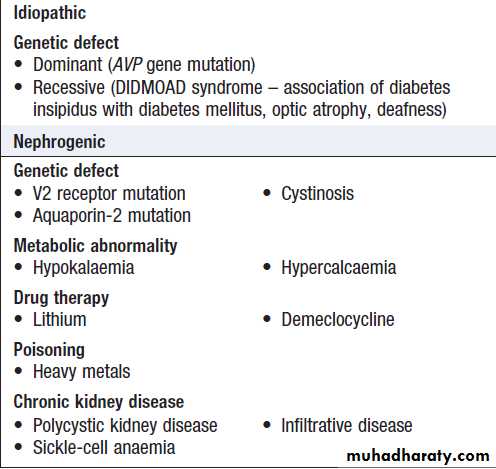
Causes of diabetes insipidus
Clinical featuresThe most marked symptoms are polyuria and polydipsia.
The patient may pass 5–20 L or more of urine in
24 hours. This is of low specific gravity and osmolality.
If the patient has an intact thirst mechanism, is conscious
and has access to oral fluids, then he or she can maintain
adequate fluid intake. However, in an unconscious patient or a patient with damage to the hypothalamic thirst centre, diabetes insipidus is potentially lethal. If there is associated cortisol deficiency, then diabetes insipidus may not be manifest until glucocorticoid replacement therapy is given. The most common differential diagnosis is primary polydipsia, caused by drinking excessive amounts of fluid in the absence of a defect in ADH or thirst control.
Investigations
Diabetes insipidus can be confirmed if serum ADH isundetectable (although the assay for this is not widely
available) or the urine is not maximally concentrated (i.e.
is below 600 mOsm/kg) in the presence of increased
plasma osmolality (i.e. greater than 300 mOsm/kg).
Sometimes, the diagnosis can be confirmed or refuted by
random simultaneous samples of blood and urine, but
more often a dynamic test is required. The water deprivation test is widely used, but an alternative is to infuse hypertonic (5%) saline and measure ADH secretion in response to increasing plasma osmolality. Thirst can also be assessed during these tests on a visual analogue scale.
Anterior pituitary function and suprasellar anatomy should be assessed in patients with cranial diabetes insipidus.
In primary polydipsia, the urine may be excessively
dilute because of chronic diuresis, which ‘washes out’
the solute gradient across the loop of Henle, but plasma
osmolality is low rather than high. DDAVP (see below)
should not be administered to patients with primary
polydipsia, since it will prevent excretion of water and
there is a risk of severe water intoxication if the patient
continues to drink fluid to excess.
In nephrogenic diabetes insipidus, appropriate
further tests include plasma electrolytes, calcium and
investigation of the renal tract .
How and when to do a water
deprivation test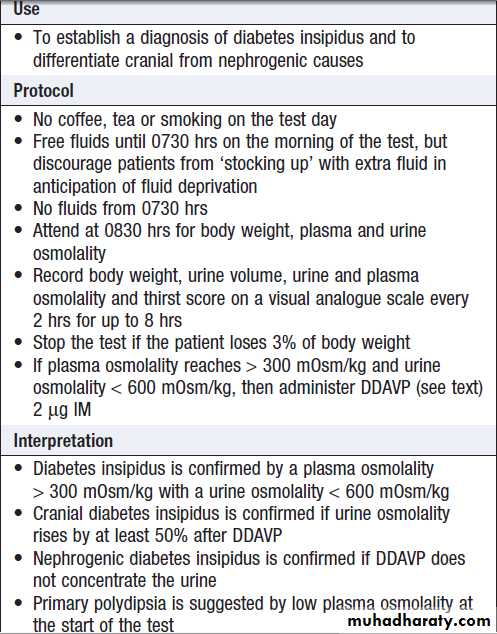
Management
Treatment of cranial diabetes insipidus is with desamino-des-aspartate-arginine vasopressin (desmopressin,
DDAVP), an analogue of ADH which has a longer half-life. DDAVP is usually administered intranasally.
An oral formulation is also available but bioavailability
is low and rather unpredictable. In sick patients, DDAVP should be given by intramuscular injection.
The dose of DDAVP should be adjusted on the
basis of serum sodium concentrations and/or osmolality.
The principal hazard is excessive treatment, resulting
in water intoxication and hyponatraemia. Conversely,
inadequate treatment results in thirst and polyuria.
The ideal dose prevents nocturia but allows a degree of polyuria from time to time before the next dose (e.g. DDAVP nasal dose 5 μg in the morning and 10 μg at night).
The polyuria in nephrogenic diabetes insipidus is improved by thiazide diuretics (e.g. bendroflumethiazide 5–10 mg/day), amiloride (5–10 mg/day) and NSAIDs (e.g. indometacin 15 mg 3 times daily), although the last of these carries a risk of reducing glomerular filtration rate.

The pituitary and hypothalamus in old age
DISORDERS AFFECTING MULTIPLE ENDOCRINE GLANDSMultiple endocrine neoplasia
Multiple endocrine neoplasias (MEN) are rare autosomal
dominant syndromes characterised by hyperplasia
and formation of adenomas or malignant tumours in
multiple glands. They fall into two groups, as shown
in Box. Some other genetic diseases also have
an increased risk of endocrine tumours; for example,
phaeochromocytoma is associated with von Hippel–
Lindau syndrome and neurofibromatosis type 1.
The MEN syndromes should be considered in all
patients with two or more endocrine tumours and inpatients with solitary tumours who report other endocrine
tumours in their family. Inactivating mutations in
MENIN, a tumour suppressor gene on chromosome 11,
cause MEN 1, whereas MEN 2 is caused by gain- offunction
mutations in the RET proto-oncogene on chromosome
10. These cause constitutive activation of the
membrane-associated tyrosine kinase RET, which controls
the development of cells that migrate from the
neural crest. In contrast, loss-of-function mutations of
the RET kinase cause Hirschsprung’s disease .
Genetic testing can be performed on relatives of affected
individuals, after appropriate counselling .
Individuals who carry mutations associated with
MEN should be entered into a surveillance programme.
In MEN 1, this typically involves annual history,
examination and measurements of serum calcium,
gastrointestinal hormones and prolactin; MRI of the pituitary and pancreas is performed at less frequent intervals. In individuals with MEN 2, annual history, examination and measurement of serum calcium and urinary catecholamine metabolites should be performed. Because the penetrance of medullary carcinoma of the thyroid is 100% in individuals with a RET mutation, prophylactic thyroidectomy should be performed in early childhood.
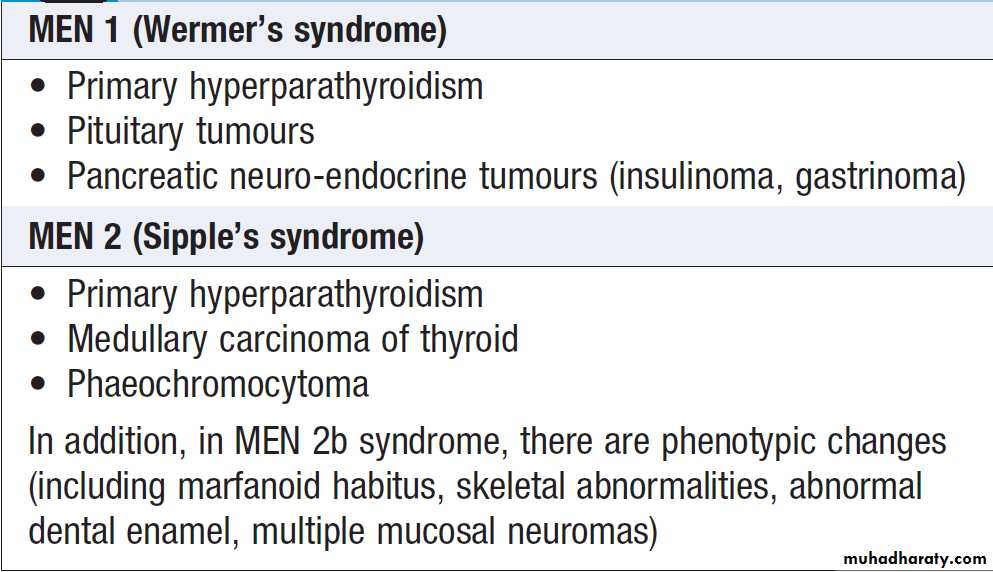
Multiple endocrine neoplasia (MEN) syndromes
Autoimmune polyendocrine syndromesTwo distinct autoimmune polyendocrine syndromes are
known: APS types 1 and 2. The most common is APS type 2 (Schmidt’s syndrome), which typically presents in women between the ages of 20 and 60. It is usually defined as the occurrence in the same individual of two or more autoimmune endocrine disorders, some of which are listed in Box . The mode of inheritance is autosomal dominant
with incomplete penetrance and there is a strong association with HLA-DR3 and CTLA-4.
Much less common is APS type 1, which is also termed autoimmune poly- endocrinopathy-candidiasis-ectodermal dystrophy (APECED).
This is inherited in an autosomal recessive fashion and is caused by loss-of function mutations in the autoimmune regulator gene AIRE, which is responsible for the presentation of self-antigens to thymocytes in utero. This is essential for the deletion of thymocyte clones that react against self-antigens and hence for the development of immune tolerance .
The most common clinical features are described in Box although the pattern of presentation is variable and other autoimmune disorders are often observed.
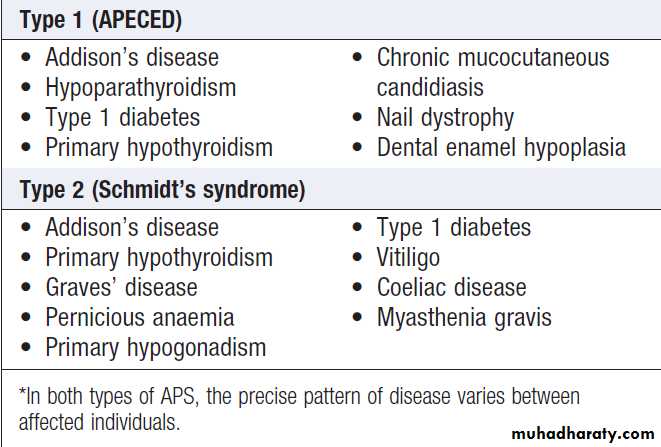
Autoimmune polyendocrine syndromes (APS)*
Late effects of childhood cancer therapyProlonged survival is increasingly common following successful treatment of cancers in children and adolescents.
The therapies used to treat these diseases, including radiotherapy and chemotherapy, may cause long-term endocrine dysfunction. In many circumstances this is predictable, such as cytotoxic chemotherapy
causing future infertility and pubertal delay (especially in boys), cranial irradiation causing long-term pituitary dysfunction, and radiotherapy to the neck causing hypothyroidism and thyroid cancer. Increasing recognition of these issues has resulted in active monitoring programmes for survivors of childhood cancer, who are best seen in specialist ‘late effects’ multidisciplinary clinics where teams include endocrinologists, oncologists, reproductive medicine specialists, psychologists and nurse specialists.