Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine

learning-topics
Rheumatology andbone disease

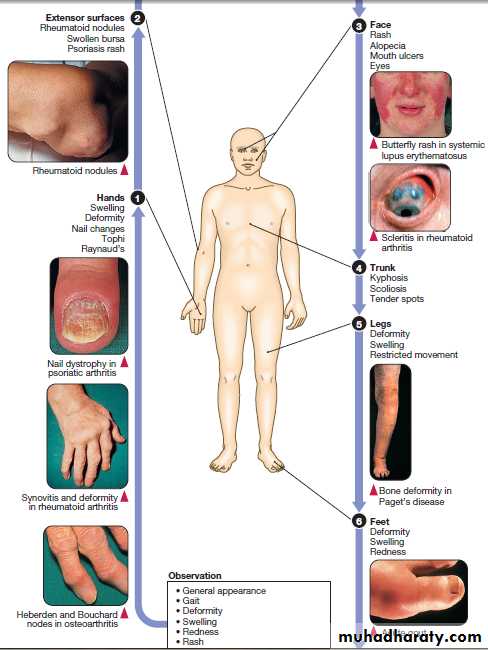
CLINICAL EXAMINATION OF THE MUSCULOSKELETAL SYSTEM
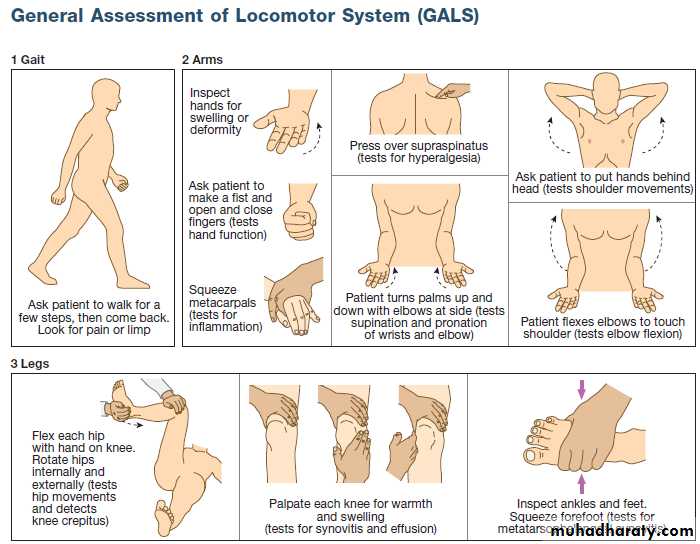
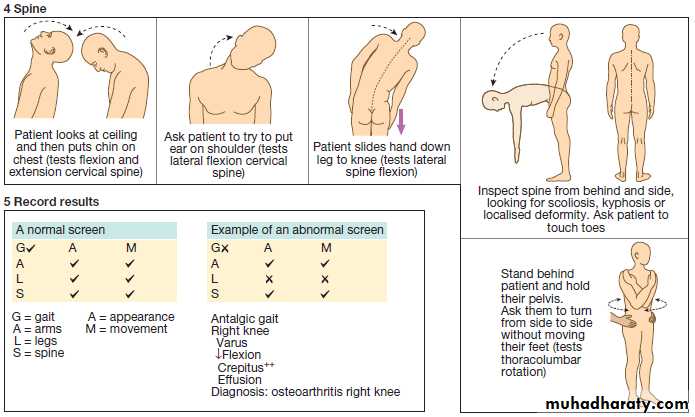
Disorders of the musculoskeletal system affect all ages .
In the UK, about 25% of new consultations in general practice are for musculoskeletal symptoms. Arise from processes affecting bones, joints, muscles, or connective tissues such as skin and tendon. The principal manifestations are pain and impairment of locomotor function. It is more common in women and increase in frequency with increasing age. The two most common disorders are osteoarthritis and osteoporosis . Osteoarthritis is the most common type of arthritis and affects up to 80% of people over the age of 75. Osteoporosis is the most common bone disease and affects 50% of women and 20% of men by their eighth decade. Account for one-third of physical disability at all ages.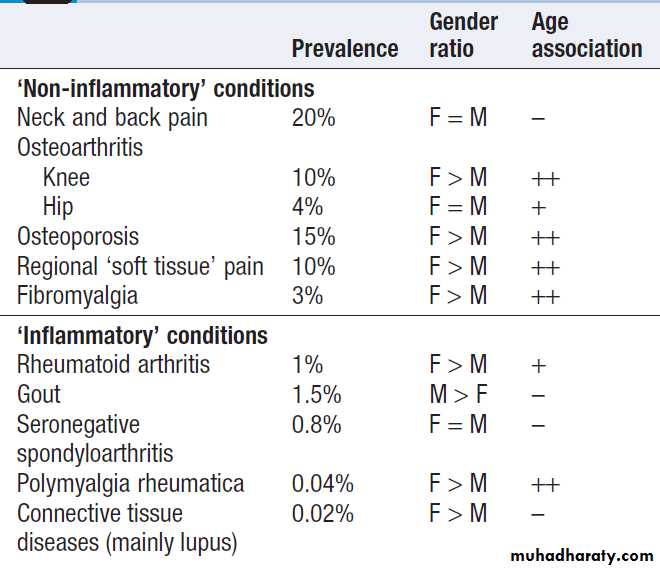
Relative prevalence of musculoskeletal disorders
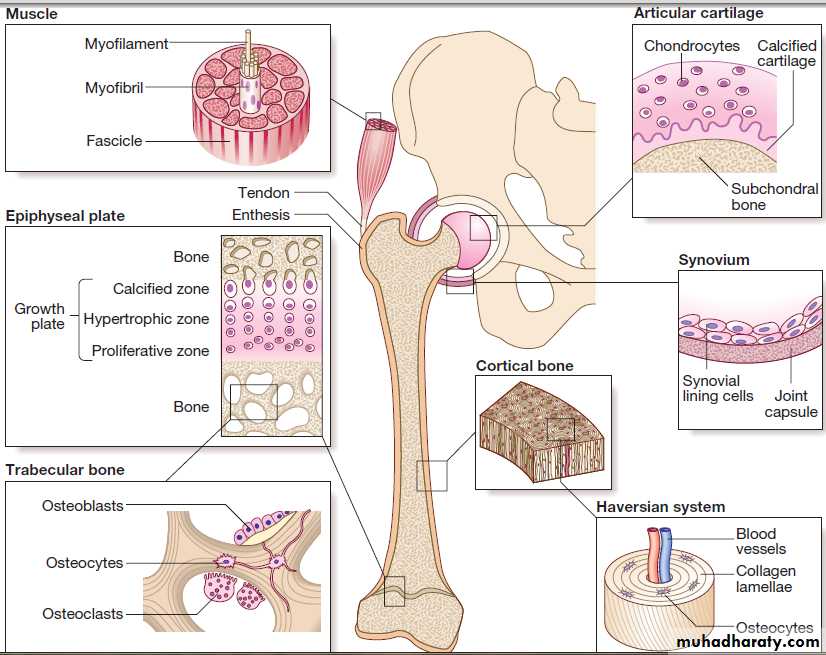
Structure of the major musculoskeletal tissues.
FUNCTIONAL ANATOMY AND PHYSIOLOGYThe musculoskeletal system is responsible for movement of the body, protect internal organs, and acts as a reservoir for storage of calcium and phosphate in the regulation of mineral homeostasis.
Bone
Bones fall into two main types.
Flat bones, such as the skull, develop by intramembranous ossification, in which embryonic fibroblasts differentiate directly into bone within condensations of mesenchymal tissue during early fetal life.
Long bones, such as the femur and radius, develop
by endochondral ossification from a cartilage template.
During development, the cartilage is invaded by vascular
tissue containing osteoprogenitor cells and is gradually
replaced by bone from centres of ossification
situated in the middle and at the ends of the bone.
A thin remnant of cartilage called the growth plate or
epiphysis remains at each end of long bones, and
chondrocyte proliferation here is responsible for skeletal
growth during childhood and adolescence.
During puberty, the rise in levels of sex hormones halts cell division in the growth plate.
The cartilage remnant then disappears as the epiphysis fuses and longitudinal bone growth ceases.
The normal skeleton has two forms of bone tissue .
Cortical bone is formed from Haversian systems, comprising concentric lamellae of bone tissuesurrounding a central canal that contains blood vessels.
Cortical bone is dense and forms a hard envelope around
the long bones.
Trabecular or cancellous bone fills the centre of the bone and consists of an interconnecting meshwork of trabeculae, separated by spaces filled with bone marrow.
There are three main cell types in bone:
• Osteoclasts: multinucleated cells of haematopoieticorigin, responsible for bone resorption.
• Osteoblasts: mononuclear cells of mesenchymal
origin, responsible for bone formation.
• Osteocytes: these differentiate from osteoblasts during bone formation and become embedded in bone matrix. Osteocytes are responsible for sensing and responding to mechanical loading of the skeleton and play a critical role in regulating bone formation and bone resorption.
They also play a central role in regulating phosphate metabolism by producing the hormone fibroblast growth factor 23 (FGF23), which acts on the kidney to promote phosphate excretion .
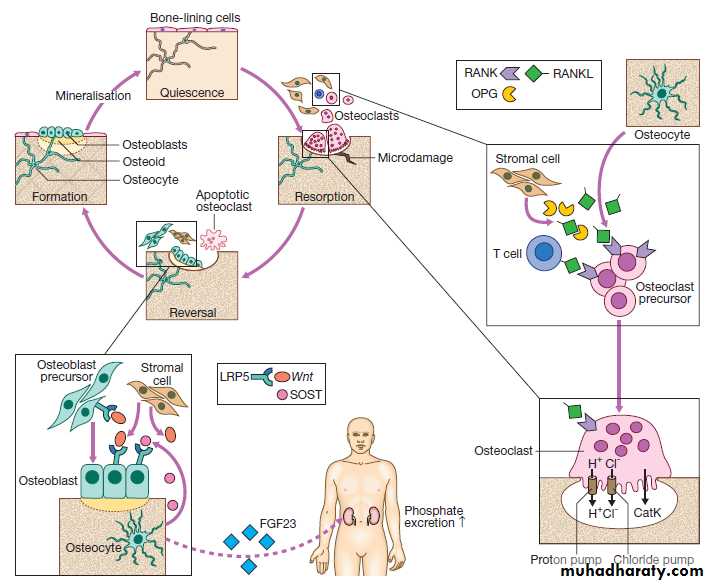
Fig. Regulation of bone remodelling. Bone is renewed and repaired during the bone remodelling cycle, in which old and damaged bone is removed by osteoclasts and replaced by osteoblasts. Osteocytes play a central role in bone remodelling by secreting RANKL, which promotes osteoclast differentiation and activity by binding to RANK. Osteocytes regulate bone formation by producing SOST, which binds to the LRP5 receptor and prevents its activation by members of the Wnt family.
Osteocytes also regulate phosphate homeostasis by producing fibroblast growth factor 23 (FGF23), which is a circulating hormone that acts on the kidney to promote phosphate excretion. (CatK = cathepsin K; LRP5 = lipoprotein receptor protein 5; OPG = osteoprotegerin; RANK = receptor activator of nuclear factor kappa B; RANKL = RANK ligand; SOST = sclerostin).
Bone matrix and mineral
The most abundant protein of bone is type I collagen,which is formed from two α1 peptide chains and one α2
chain wound together in a triple helix. Type I collagen
is proteolytically processed inside the cell before being
laid down in the extracellular space, releasing propeptide
fragments that can be used as biochemical markers
of bone formation .
Subsequently, the collagen fibrils become ‘cross-linked’ to one another by pyridinium molecules, a process that enhances bone strength.
When bone is broken down by osteoclasts, the crosslinks are released, providing biochemical markers of bone resorption.
Bone is normally laid down in an orderly fashion, but when bone turnover is high, as in Paget’s disease or severe hyperparathyroidism, it is laid down in a chaotic pattern, giving rise to ‘woven bone’, which is mechanically weak. Bone matrix also contains growth factors, other structural proteins and proteoglycans, thought to be involved in helping bone cells attach to bone matrix and in regulating bone cell activity. The other major component of bone is mineral, comprised of calcium and phosphate crystals deposited between the collagen fibrils in the form of hydroxyapatite [Ca10 (PO4)6 (OH)2]. Mineralisation is essential for bone’s rigidity and strength, but over-mineralisation can increase brittleness, which contributes to bone fragility in diseases like osteogenesis imperfecta .
Bone remodelling
Bone remodelling is required for renewal and repair ofthe skeleton throughout life . It starts with the attraction of osteoclast precursors in peripheral blood to the target site, probably by local release of chemotactic factors from areas of microdamage.
The osteoclast precursors differentiate into mature osteoclasts in response to RANKL, which is produced by
osteocytes, activated T cells and bone marrow stromal
cells. RANKL activates the RANK receptor, which is
expressed on osteoclasts and precursors. This is blocked
by osteoprotegerin (OPG), a decoy receptor for RANKL
that inhibits osteoclast formation.
Mature osteoclasts attach to the bone surface by a tight sealing zone, and secrete hydrochloric acid and proteolytic enzymes such as cathepsin K into the space underneath. The acid dissolves the mineral and cathepsin K degrades collagen.
When resorption is complete, osteoclasts undergo programmed cell death, and bone formation begins with
the attraction of osteoblast precursors to the resorption
site. These differentiate into mature osteoblasts, which
deposit new bone matrix in the resorption lacuna, until
the hole is filled. Some osteoblasts become trapped in
bone matrix and differentiate into osteocytes. These act as biomechanical sensors and produce several molecules that influence bone remodelling and phosphate metabolism.
Bone formation is stimulated by Wnt proteins, which bind to and activate lipoprotein-related receptor protein 5 (LRP5), expressed on osteoblasts. This process is inhibited by SOST, which is produced by osteocytes .
Initially, the newly formed bone matrix (osteoid) is uncalcified but subsequently becomes mineralized to form mature bone. Alkaline phosphatase (ALP), produced by osteoblasts, plays an important role in bone mineralisation by degrading pyrophosphate, an inhibitor of mineralisation.
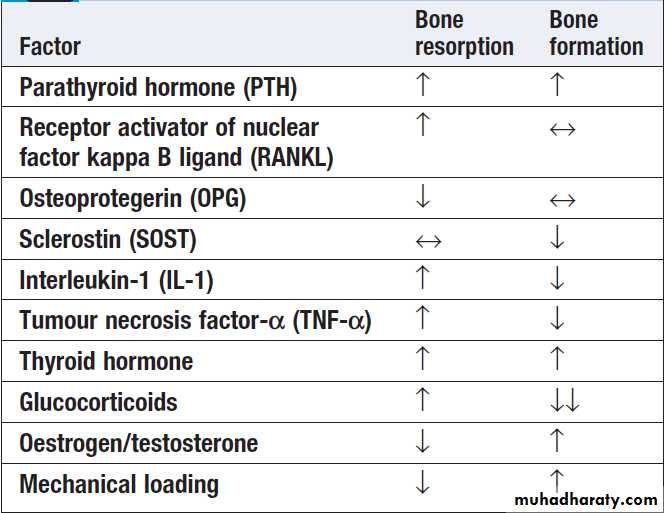
Bone remodelling is regulated by circulating hormones such as parathyroid hormone (PTH) and oestrogen, and locally produced factors such as cytokines .
Regulators of bone remodelling
Types of joint
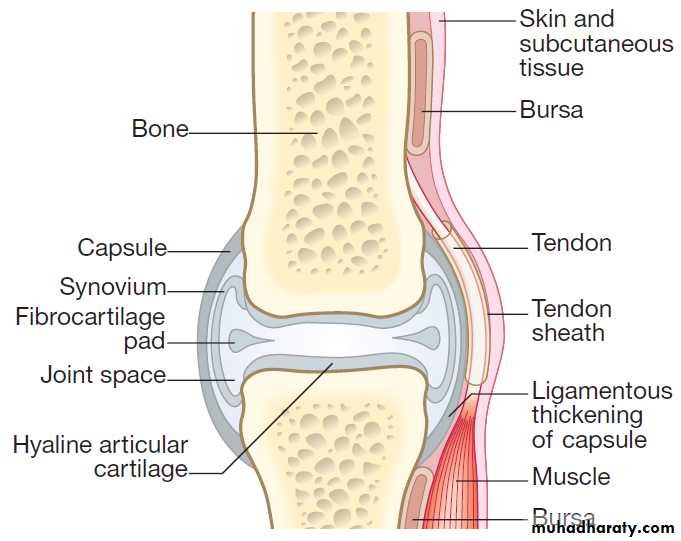
Structure of a synovial joint
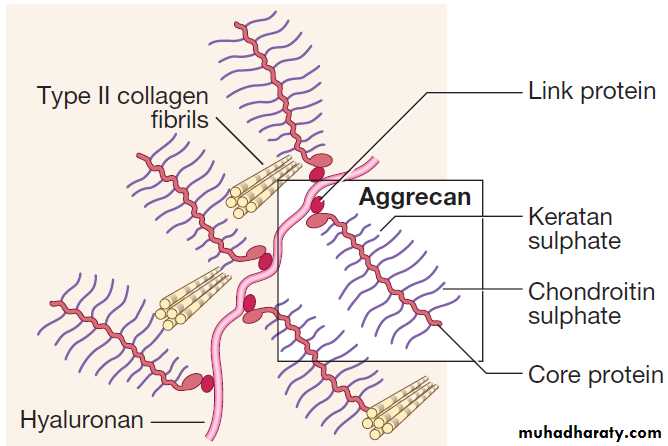
Ultrastructure of articular cartilage
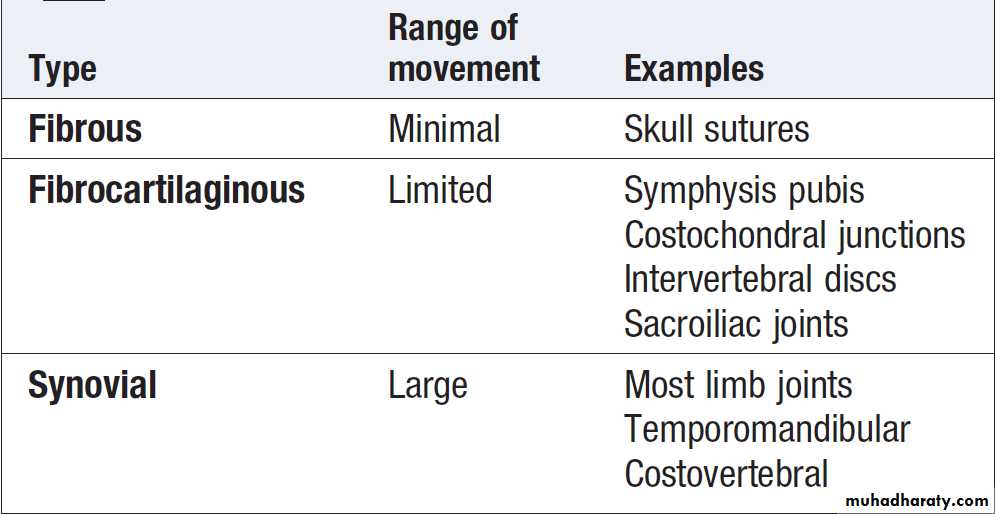
JointsBones are linked by joints. There are three main subtypes:
fibrous, fibrocartilaginous and synovial .
Fibrous and fibrocartilaginous joints
These comprise a simple bridge of fibrous or fibrocartilaginous tissue joining two bones together where there is little requirement for movement. The intervertebral disc is a special type of fibrocartilaginous joint in which an amorphous area, called the nucleus pulposus, lies in the centre of the fibrocartilaginous bridge. The nucleus has a high water content and acts as a cushion to improve the disc’s shock-absorbing properties.
Synovial joints
Complex structures containing several cell types, theseare found where a wide range of movement is needed.
Articular cartilage
This avascular tissue covers the bone ends in synovial
joints. Cartilage cells (chondrocytes) are responsible for
synthesis and turnover of cartilage, which consists of a
mesh of type II collagen fibrils that extend through a
hydrated ‘gel’ of proteoglycan molecules. The most important proteoglycan is aggrecan, which consists of a
core protein to which several glycosaminoglycan (GAG)
side-chains are attached . The expansive force of the hydrated aggrecan, combined with the restrictive strength of the collagen mesh, gives articular cartilage excellent shock-absorbing properties.
With ageing, the concentration of chondroitin sulphate
decreases, whereas that of keratan sulphate
increases, resulting in reduced water content and shockabsorbing properties. These changes differ from those found in osteoarthritis , where there is abnormal
chondrocyte division, loss of proteoglycan from matrix
and an increase in water content. Cartilage matrix is
constantly turning over and in health there is a perfect
balance between synthesis and degradation. Pro-inflammatory cytokines, such as interleukin-1 (IL-1)
and tumour necrosis factor (TNF), stimulate production
of aggrecanase and metalloproteinases, which contribute
to cartilage degradation in inflammatory arthritis.
Synovial fluid
The surfaces of articular cartilage are separated bya space filled with synovial fluid, a viscous liquid
that lubricates the joint. It is an ultrafiltrate of plasma,
into which synovial cells secrete hyaluronan and
proteoglycans.
Intra-articular discs
Some joints contain fibrocartilaginous discs within the
joint space that act as shock absorbers. The most clinically
important are the menisci of the knee. These are
avascular structures that remain viable by diffusion of
oxygen and nutrients from the synovial fluid.
The synovial membrane, joint capsule and bursae
The bones of synovial joints are connected by the
joint capsule, a fibrous structure richly supplied with
blood vessels, nerves and lymphatics, which encases the
joint. Ligaments are discrete, regional thickenings of
the capsule that act to stabilise joints . The inner surface of the joint capsule is the synovial membrane, comprising an outer layer of blood vessels and loose connective tissue that is rich in type I collagen, and an inner layer 1–4 cells thick consisting of two main cell types.
Type A synoviocytes are phagocytic cells derived from the monocyte/macrophage lineage and are responsible for removing particulate matter from the joint cavity; type B synoviocytes are fibroblast-like cells that secrete synovial fluid. Most inflammatory and degenerative joint diseases associate with thickening of the synovial membrane and infiltration by lymphocytes, polymorphs and macrophages.
Bursae are hollow sacs lined with synovium and
contain a small amount of synovial fluid.
They help tendons and muscles move smoothly in relation to bones and other articular structures.
Skeletal muscle
Skeletal muscles are responsible for body movementsand respiration. Muscle consists of bundles of cells
(myocytes) embedded in fine connective tissue containing
nerves and blood vessels. Myocytes are large,
elongated, multinucleated cells formed by fusion of
mononuclear precursors (myoblasts) in early embryonic
life. The nuclei lie peripherally and the centre of the cell
contains actin and myosin molecules, which interdigitate
with one another to form the myofibrils that are
responsible for muscle contraction.
The molecular Mechanisms of skeletal muscle contraction are the same as for cardiac muscle .
Myocytes contain many mitochondria that provide the large amounts of adenosine triphosphate (ATP) necessary for muscle contraction, and are rich in the protein myoglobin, which acts as a reservoir for oxygen during contraction.
Tendons are tough, fibrous structures that attach muscles to a point of insertion on the bone surface called the enthesis.
INVESTIGATION OF MUSCULOSKELETAL DISEASE
Clinical history and examination usually provide sufficientinformation for the diagnosis and management of most musculoskeletal diseases. Investigations are helpful in confirming the diagnosis, assessing disease activity and indicating prognosis.
Joint aspiration
Joint aspiration with examination of synovial fluid (SF)
is pivotal in patients suspected of having septic arthritis,
crystal arthritis or intra-articular bleeding. It should be
done in all patients with acute monoarthritis, and samples sent for microbiology and clinical chemistry. It is possible to obtain SF by aspiration from most peripheral joints, and only a small volume is required for diagnostic purposes.
Normal SF is present in small volume, and is clear and either colourless or pale yellow with a high viscosity. It contains few cells. With joint inflammation, the volume increases, the cell count and the proportion of neutrophils rise (causing turbidity), and the viscosity reduces (due to enzymatic degradation of hyaluronan and aggrecan).
Turbid fluid with a high neutrophil count occurs in sepsis, crystal arthritis and reactive arthritis. High concentrations of urate crystals or cholesterol can make SF appear white. Non-uniform blood-staining usually reflects needle trauma to the synovium. Uniform blood-staining is most commonly due to a bleeding diathesis, trauma or pigmented villonodular synovitis , but can occur in severe synovitis.
A lipid layer floating above blood-stained
fluid is diagnostic of intra-articular fracture and iscaused by release of bone marrow fat into the joint.
Crystals can be identified by compensated polarised
light microscopy of fresh SF (to avoid crystal dissolution
and post-aspiration crystallisation). Urate crystals are
long and needle-shaped, and show a strong light intensity
and negative birefringence . Calcium pyrophosphate crystals are smaller, rhomboid in shape
and usually less numerous than urate, and have weak
intensity and positive birefringence .
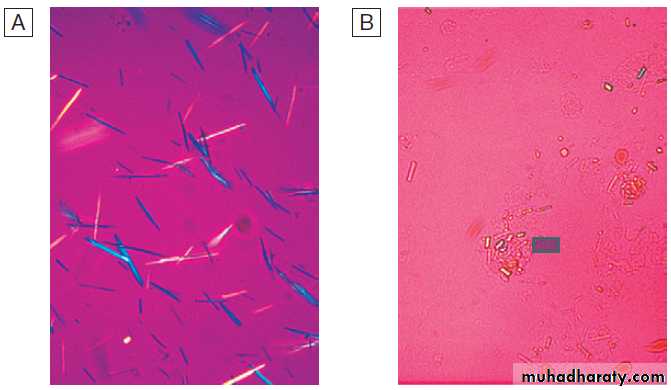
Compensated polarised light microscopy of synovial
fluids (× 400).A Monosodium urate crystals showing bright negative birefringence under polarised light and needle-shaped morphology.
B Calcium pyrophosphate crystals showing weak positive birefringence under polarised light and are few in number. They are more difficult to
detect than urate crystals.
Imaging
Plain radiographyShow changes of many bone and joint diseases . Radiographs are of diagnostic value in osteoarthritis
(OA), where they demonstrate joint space narrowing
that tends to be focal rather than widespread, as in
inflammatory arthritis. Other features of OA detected on
X-ray include osteophytes, subchondral sclerosis, bone
cysts and calcified loose bodies within the synovium . Radiographs may show erosions and sclerosis of the sacroiliac joints and syndesmophytes in the spine in seronegative spondyloarthritis. In peripheral joints, so-called proliferative erosions, associated with new bone formation and a periosteal reaction, may be observed.
In tophaceous gout, well-defined punched-out erosions may occur .
Calcification of cartilage, tendons and soft tissues or muscle may occur in chondrocalcinosis , calcific periarthritis and connective tissue diseases. Radiographs are of limited value in the diagnosis of rheumatoid arthritis (RA) since features such as erosions, joint space narrowing and periarticular osteoporosis may only be detectable after several months or even years.Early evidence of articular damage in RA is more usually obtained using MRI or ultrasonography.
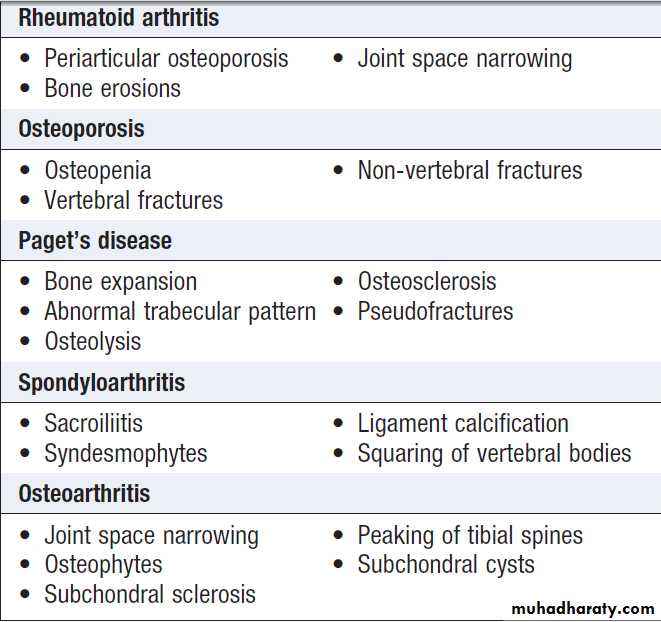
X-ray abnormalities in selected
rheumatic diseasesRadionuclide bone scan
This is useful in patients suspected of having metastatic bone disease and Paget’s disease. It involves gamma-camera imaging following an intravenous injection of 99mTc-bisphosphonate. Early post-injection images reflect vascularity and can show increased perfusion of inflamed synovium, Pagetic bone, or primary or secondary bone tumours. Delayed images taken a few hours later reflect bone remodelling as the bisphosphonate localises to sites of active bone turnover. Scintigraphy has a high sensitivity for detecting important bone and joint pathology that is not apparent on plain X-rays . Single photon emission computed tomography (SPECT) combines radionuclide imaging with computed tomography. It can provide accurate anatomical localization of abnormal tracer uptake within the bone and is of particular value in the assessment of patients with chronic low back pain of unknown cause.


Conditions detected by isotope bone scanning

Magnetic resonance imaging
Gives detailed information on anatomy, allowing three-dimensional visualization of bone and soft tissues that cannot be adequately assessed by plain X-rays. The technique is valuable in the assessment and diagnosis of many musculoskeletal diseases . T1-weighted sequences are useful for defining anatomy, whereas T2-weighted sequences are useful for assessing tissue water content, which is often increased in synovitis and other inflammatory disorders . Contrast agents, such as gadolinium, can be administered to increase sensitivity in detecting erosions and synovitis.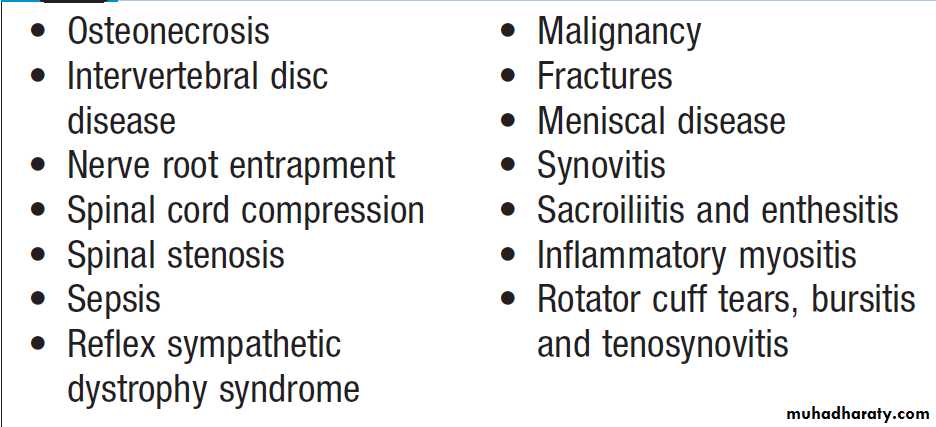
Conditions detected by magnetic resonance imaging
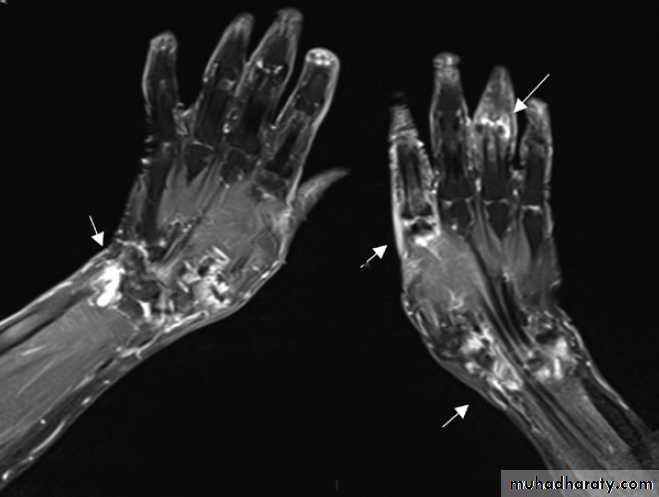
Magnetic resonance image showing synovitis. Coronal
fat-saturated post-contrast T1-weighted image shows extensiveenhancement consistent with synovitis (white areas, arrowed) in both wrists
and at the second metacarpophalangeal joint and proximal interphalangeal
joints of the right hand.
Ultrasonography
Ultrasonography is a useful investigation for confirmationof small joint synovitis and erosion, for anatomical
confirmation of periarticular lesions, and for guided
aspiration and injection of joints and bursae. Ultrasound
is more sensitive than clinical examination for the
detection of early synovitis and is used increasingly
in the diagnosis and assessment of patients with suspected
inflammatory arthritis. In addition to locating
synovial thickening and effusions, ultrasound can detect
increased blood flow within synovium using power
Doppler imaging, an option that is available on most
modern ultrasound machines .
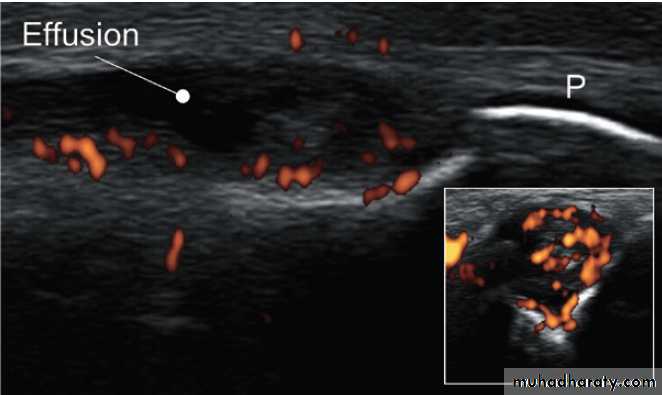
Ultrasound image showing synovitis. Lateral image of a metacarpophalangeal joint in inflammatory arthritis. The periosteum (P) of the phalanx shows as a white line. The dark, hypo-echoic area indicates
an effusion. The coloured areas demonstrated by power Doppler indicate
increased vascularity. The inset shows a transverse image of the same joint.
Computed tomography
Computed tomography (CT) can be used in the assessmentof patients with bone and joint disease but
has largely been superseded by MRI, which gives
better visualisation of soft tissue structures. CT may be
used when MRI is contraindicated, or for evaluation
of articular regions in which an adjacent joint replacement
creates image artefacts on MRI.
Bone mineral density (BMD)
BMD measurements play a key role in the diagnosis and management of osteoporosis. The technique of choice is dual energy X-ray absorptiometry (DEXA), which is usually performed at the lumbar spine and hip.This technique works on the principle that calcium in bone attenuates passage of X-ray beams through the
tissue in proportion to the amount of mineral present. The greater the amount of bone mineral present, the
higher the BMD value. Most DEXA scanners give a BMD
readout expressed as grams of hydroxyapatite/cm2, and
as a T-score and Z-score value. Osteoporosis is defined by a T-score value of 2.5 or below (shaded red in the figure), whereas osteopenia is diagnosed when the T-score lies between −1.0 and −2.5 (shaded pink).
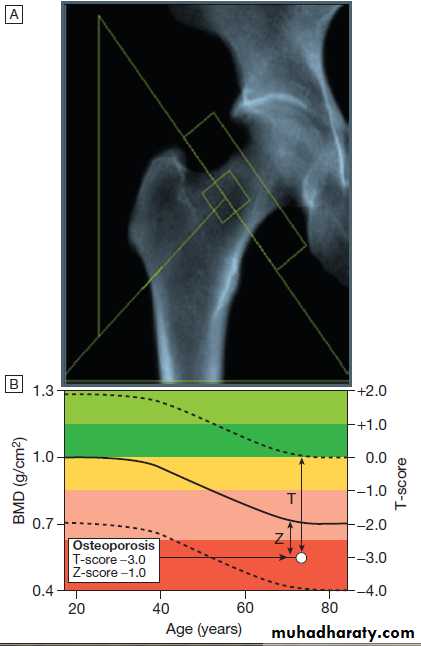
Fig. Typical output from a dual energy X-ray absorptiometry
(DEXA) scanner.A DEXA scan of the hip.
B Bone mineral density
(BMD) values plotted in g/cm2 (left axis) and as the T-score values (right axis). The solid line represents the population average plotted against age,
and the interrupted lines are ± 2 standard deviations from the average.
The patient shown, aged 72, has an osteoporotic T-score of −3.0 but a Z-score of −1.0, which is within the ‘normal range’ for that age, reflecting the fact that bone is lost with age.
Blood tests
HaematologyAbnormalities in the full blood count (FBC) often occur
in inflammatory rheumatic diseases but changes are
usually non-specific. Examples include neutrophilia in
vasculitis, acute gout and sepsis; neutropenia in lupus;
and a raised ESR in many inflammatory diseases. Reduced levels of haemoglobin are a common and important finding in a range of rheumatological disorders. Many disease-modifying antirheumatic drugs (DMARDs) cause marrow toxicity and require regular monitoring of the FBC.
Biochemistry
Routine biochemistry is useful for assessing metabolicbone disease, muscle diseases and gout. Several bone
diseases, including Paget’s disease, renal bone disease
and osteomalacia, give a characteristic pattern that can
be helpful diagnostically (Box). Serum levels of uric
acid are usually raised in gout but a normal level does
not exclude it, especially during an acute attack, when
urate levels temporarily fall. Equally, an elevated serum
uric acid does not confirm the diagnosis, since most
Hyperuricaemic people never develop gout. Levels of
C-reactive protein (CRP) are a useful marker of infection
and inflammation, and are more specific than the ESR.
An exception is in connective tissue diseases such as
systemic lupus erythematosus (SLE) and systemic sclerosis,
where CRP may be normal but ESR raised in active
inflammatory disease. Accordingly, an elevated CRP in
a patient with lupus or scleroderma suggests an Intercurrent illness such as sepsis rather than active disease.
Serum creatine kinase (CK) levels are useful in the diagnosis of myopathy or myositis, but specificity and sensitivity are poor and raised levels may occur in some conditions . Biochemical monitoring of renal and hepatic function is important in patients on DMARD therapy.
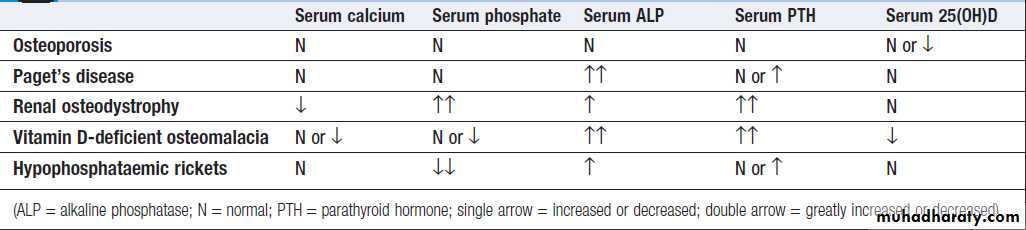
Biochemical abnormalities in bone disease

Causes of an elevated serum creatine kinase
AutoantibodiesAutoantibody tests are widely used in the diagnosis of
rheumatic diseases. False-positive results are common
but high antibody titres or concentrations are generally
of greater clinical significance. Whatever test is used, the results must be interpreted in light of the clinical picture. Rheumatoid factor (RF)
Antibody directed against the Fc fragment of human immunoglobulin. IgM RF is usually measured, although different methodologies allow measurement of IgG and IgA RFs too. Positive RF occurs in a wide variety of diseases and some normal particularly with increasing age. Although the specificity is poor, about 70% with RA test positive. High RF titres are associated with more severe disease and extra-articular disease.
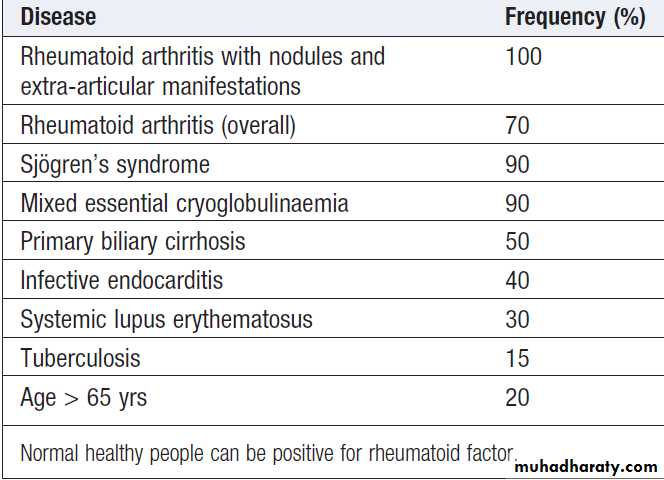
Conditions associated with a positive rheumatoid factor
Anti-citrullinated peptide antibodies (ACPA)ACPA have similar sensitivity to RF for RA (70%) but much higher specificity (> 95%), and are increasingly being used in preference to RF in the diagnosis of RA. ACPA are associated with more severe disease progression, and can be detected in asymptomatic patients several years before the development of RA. Their pathological role is still debated but it is likely that they amplify the synovial response at an inflammatory stimulus.
Antinuclear antibodies (ANA)
Directed against one or more components of the cell nucleus. They occur in many inflammatory rheumatic diseases but are also found at low titre in normal individuals and in other diseases.
High titres of ANA are of greater diagnostic significance but circulating levels are not associated with disease severity or activity. The most common indication for ANA testing is in patients suspected of SLE or connective tissue diseases. ANA has high sensitivity for SLE (virtually 100%) but low specificity (10– 40%). A negative ANA virtually excludes SLE but a positive result does not confirm it. Anti-DNA antibodies bind to double-stranded DNA and are highly specific for SLE (95%) but sensitivity is poor (30%). They can be useful in disease monitoring
since very high titres are associated with more severe
disease. Antibodies to extractable nuclear antigens (ENA) act as markers for certain CT diseases and some complications of SLE, but poor sensitivity and specificity.
For example, antibodies to Sm are found in a minority of SLE patients but are associated with renal involvement.
Antibodies to Ro occur in SLE and in Sjِgren’s syndrome (in association with anti-La antibodies), and are associated with a photosensitive rash and congenital heart block. Antibodies to ribonuclear protein (RNP) occur in SLE
and also in mixed connective tissue disease, where
features of lupus, myositis and systemic sclerosis
coexist.
Anti-topoisomerase 1 (also termed Scl-70) antibodies occur in diffuse systemic sclerosis, whereas anti-centromere antibodies are more specific for limited systemic sclerosis.
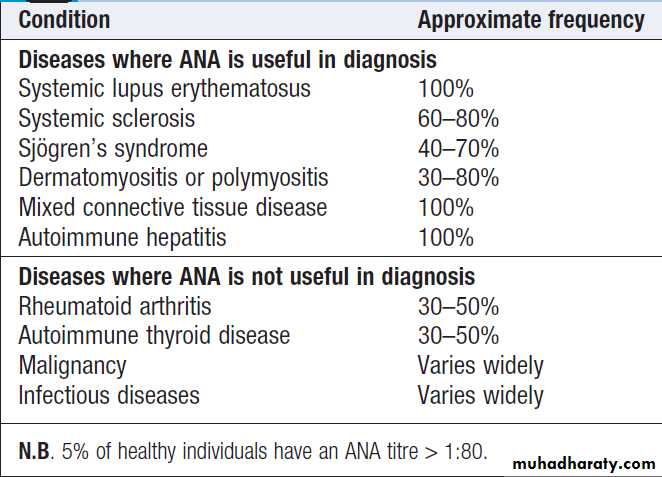
Conditions associated with a positive antinuclear antibody
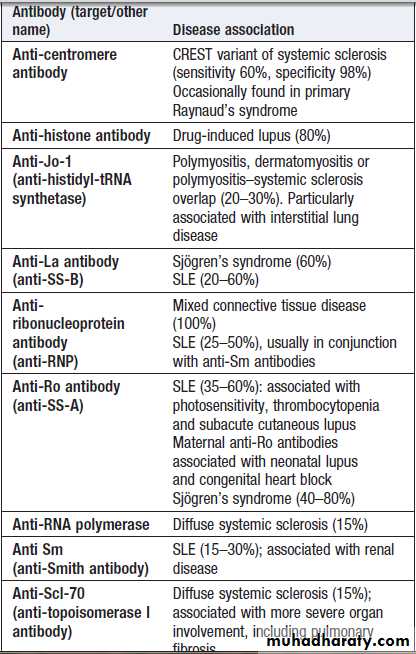
Conditions associated with antibodies to
extractable nuclear antigensAntiphospholipid antibodies
Bind to a number of phospholipid binding proteins, but the most clinically relevant are those that target beta2-glycoprotein 1 (β2GP1). They may be detected in SLE and other CT diseases or can be present in isolation or in the antiphospholipid antibody syndromeAntineutrophil cytoplasmic antibodies
Antineutrophil cytoplasmic antibodies (ANCA) are IgG
antibodies directed against the cytoplasmic constituents
of granulocytes and are useful in the diagnosis and
monitoring of systemic vasculitis. These antibodies are not specific for vasculitis and positive results may be found in autoimmune liver disease, malignancy, infection (bacterial and HIV), inflammatory bowel disease, rheumatoid arthritis, SLE and pulmonary fibrosis.
Tissue biopsy
Synovial biopsy can be useful in chronic inflammatory monoarthritis or tenosynovitis to rule out chronic infectious causes, especially mycobacterial infections. Characteristic changes on MRI can obviate the need for biopsy in many cases of suspected tumour. synovial biopsy can be obtained arthroscopically (by conventional meansor via use of needle arthroscope) or using ultrasound
guidance under local anaesthetic. Temporal artery biopsy can be of value in suspected of having temporal arteritis, but a negative result does not exclude the diagnosis. Biopsies of affected tissues, such as skin, lung, nasopharynx, gut, kidney and muscle, can be of value in the diagnosis of systemic vasculitis and granulomatosis with polyangiitis (Wegener’s granulomatosis).
Muscle biopsy plays an important role in the investigation
of myopathy and inflammatory myositis. It is usually taken from the quadriceps or deltoid through a small skin incision under local anaesthetic. Since myositis can be patchy in nature, MRI is sometimes used to localise the best site. Repeat biopsies are sometimes used to monitor the response to treatment. Bone biopsy is occasionally required where noninvasive tests give inconclusive and in the diagnosis of infiltrative disorders, chronic infections and malignancy. If osteomalacia, is suspected, the biopsy can be taken from the iliac crest using trephine needle under local anaesthetic. For focal lesions, the biopsy
should be taken under X-ray guidance or at open
surgery, from an affected site.
Electromyography
Electromyography is of value in the investigationof suspected myopathy and inflammatory myositis,
when it shows the diagnostic triad of:
• spontaneous fibrillation
• short-duration action potentials in a polyphasic
disorganised outline
• repetitive bouts of high-voltage oscillations on
needle contact with diseased muscle.
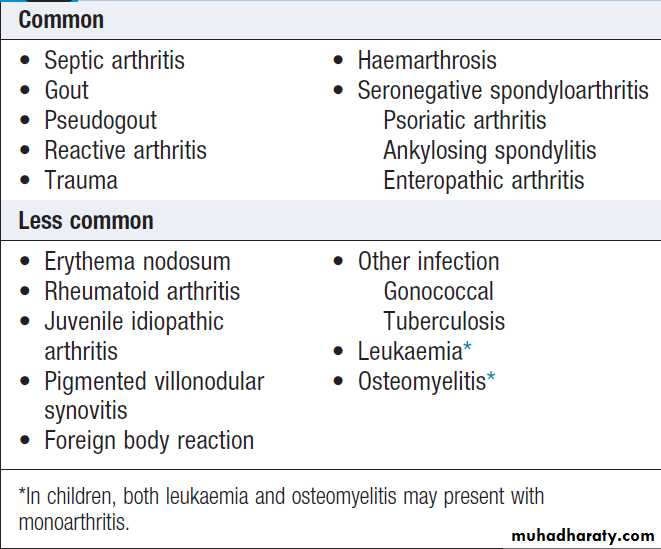
Causes of acute monoarthritis
PRESENTING PROBLEMS IN MUSCULOSKELETAL DISEASEAcute monoarthritis
Sudden pain and swelling in a single joint. The most important are crystal, sepsis and reactive arthritis.
Clinical assessment
The clinical history, pattern of joint involvement, speed
of onset, and age and gender of the patient all give clues
to the most likely diagnosis. Reactive arthritis is the most common cause in young men, gout in middle-aged
men and pseudogout in older women.
Gout classically affects the first metatarsophalangeal (MTP) joint, whereas the wrist and shoulder are typical sites for pseudogout.
A very rapid onset (6–12 hours) is suggestive of gout or pseudogout; joint sepsis develops more slowly and continues to progress until treated. Haemarthrosis typically causes a large effusion, in the absence of periarticular swelling or skin change.A previous diarrhoeal illness or recent sexual contact suggests reactive, whereas intercurrent illness, dehydration or surgery may act as a trigger for crystal-induced arthritis.
Rheumatoid arthritis seldom presents with monoarthritis
and a sudden increase in pain and swelling involving
a single joint in pre-existing RA is strongly suggestive of sepsis. Osteoarthritis can present with pain and stiffness affecting a single joint, but the onset is gradual and there is seldom evidence of significant joint swelling.
Investigations
Aspiration of the affected joint is mandatory. The fluidshould be sent for culture and Gram stain to seek the
presence of organisms, and should be checked by microscopy for crystals. Blood cultures should also be
taken in patients suspected of having septic arthritis.
CRP levels and ESR are raised in sepsis, crystal arthritis
and reactive arthritis, and this can be useful in assessing
the response to treatment.
Serum uric acid measurements may be raised in gout but a normal level does not exclude the diagnosis.
Management
If there is any suspicion of sepsis,IV antibiotics should be given promptly, pending the results of cultures. Otherwise, management should be directed towards the cause.
Causes of polyarthritis
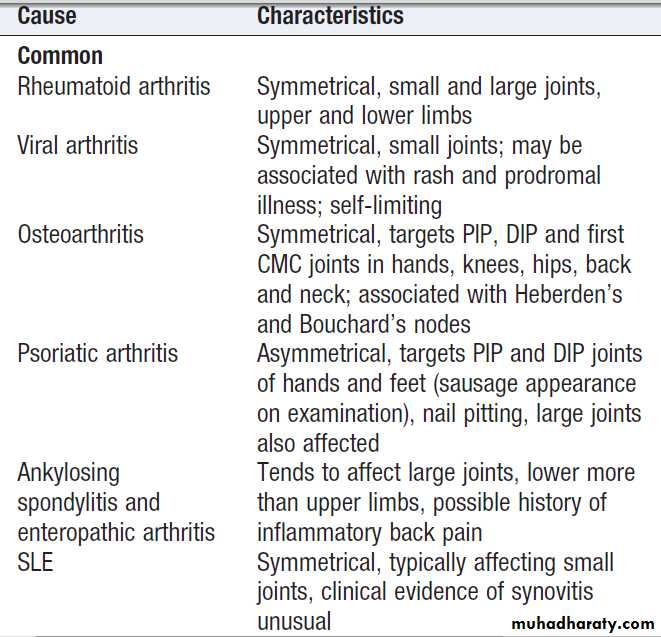
Causes of polyarthritis – cont’d
Less common
Juvenile idiopathic arthritis
Symmetrical, small and large joints, upper and lower limbs
Chronic gout Affects distal more than proximal joints, history of acute attacks
Chronic sarcoidosis
Symmetrical, small and large joints
Polymyalgia rheumatic
Symmetrical, small and large joints
Rare
Systemic sclerosis and polymyositis
Small and large joints
Hypertrophic osteoarthropathy
Small joints, clubbing
Haemochromatosis
Small and large joints
Acromegaly
Mainly large joints and spine
Polyarthritis
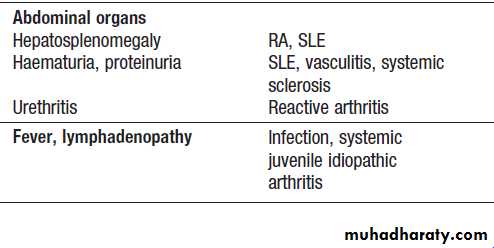
This term is used to describe pain and swelling affecting five or more joints or joint groups.Clinical assessment
The hallmarks of inflammatory arthritis are early morning stiffness and worsening of symptoms with inactivity, along with synovial swelling and tenderness on
examination. The most important diagnosis to consider is rheumatoid arthritis, which is characterised by symmetrical involvement of the small joints of the hands and feet, often in association with other joints. Viral arthritis should also be considered. This presents with an acute symmetrical inflammatory polyarthritis affecting small and large joints of upper and lower limbs, often with a rash.
The pattern of involvement can be helpful in reaching a diagnosis . Asymmetry, lower limb predominance and greater involvement of large joints are characteristic of seronegative spondyloarthritis. Other extra-articular features may also be present, giving a clue to the diagnosis. In psoriatic arthritis, the small joints of the hand and feet are often affected, with involvement of the proximal and distal interphalangeal (PIP and DIP) joints, as opposed to the MCP and PIP joints in RA.
The pattern of involvement also tends to be asymmetrical in psoriatic arthritis, and other clues such as nail pitting and a rash may be present.
SLE can be associated with polyarthritis but more usually causes polyarthralgia and tenosynovitis .
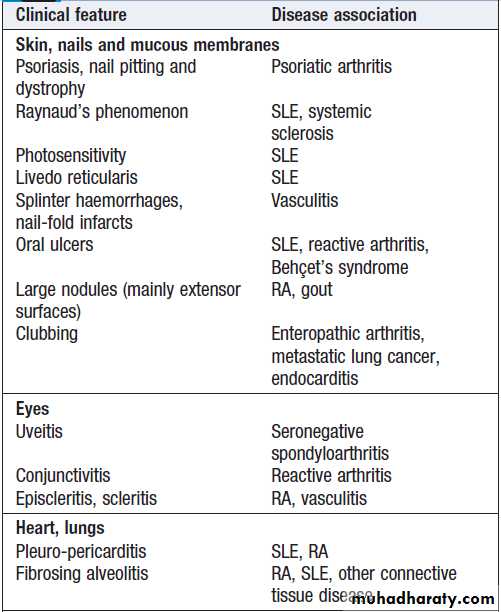
Extra-articular features of inflammatory arthritis
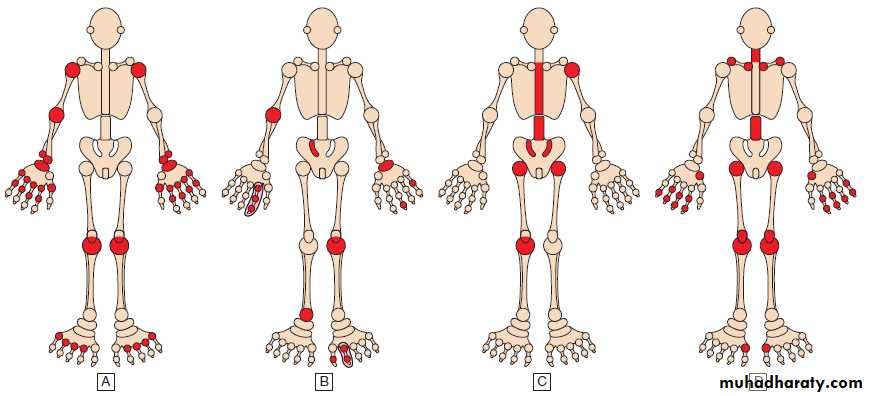
Fig. Patterns of joint involvement in different forms of polyarthritis.
A Rheumatoid arthritis typically targets the etacarpophalangeal and proximal interphalangeal joints of the hands and metatarsophalangeal joints of the feet, as well as other joints, in a symmetrical pattern.B Psoriatic arthritis targets proximal and distal interphalangeal joints of the hands and larger joints in an asymmetrical pattern. Sacroiliitis (often asymmetrical) may occur.
C Ankylosing spondylitis targets the spine, sacroiliac joints and large peripheral joints in an asymmetrical pattern.
D Osteoarthritis targets the proximal and distal interphalangeal joints of the hands, first carpometacarpal joint at the base of the thumb, knees, hips, lumbar and cervical spine.
Investigations
Blood samples should be taken for routine haematology,
biochemistry, ESR, CRP, viral serology and an
immunological screen, including ANA, RF and ACPA.
Ultrasound examination or MRI may be required to
confirm the presence of synovitis if this is not obvious
clinically.
Management
should be directed at the underlying condition, but treatment with NSAIDs and analgesics may be required for symptom control until a diagnosis has been made.
Fracture
Fractures are a common presenting symptom of osteoporosis, but they also occur in other bone diseases, in osteopenia and in some patients with normal bone.Clinical assessment
The presentation is with localised bone pain, which is worsened by movement of the affected limb or region.
There is usually a history of trauma but spontaneous fractures can occur in the absence of trauma in those with severe osteoporosis. The main differential diagnosis is soft tissue injury, but fracture should be suspected when there is marked pain and swelling, abnormal movement of the affected limb, crepitus or deformity. Femoral neck fractures typically produce a shortened, externally rotated leg that is painful to move.
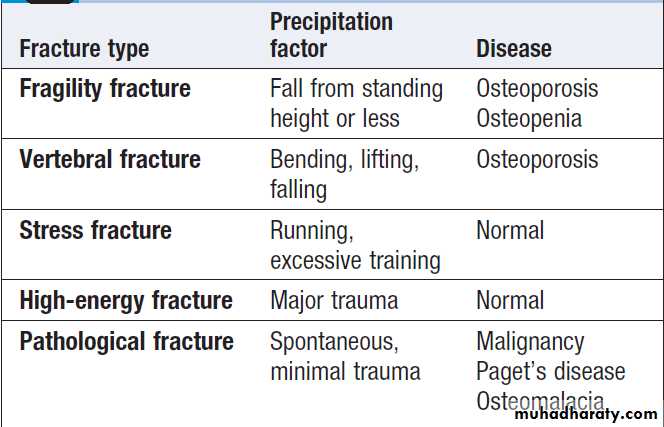
Characteristics of different fracture types

How to investigate a suspected fracture
Investigations
Radiographs of the affected site should be taken in atleast two planes and examined for discontinuity of the
cortical outline . In addition to demonstrating the fracture, X-rays may also show evidence of an underlying disorder, such as osteoporosis, Paget’s disease or osteomalacia. If the X-ray fails to show evidence of a
fracture but clinical suspicion remains high, MRI should
be performed, since this can demonstrate fractures that
are radiographically occult. Patients who are over the
age of 50 and present with fragility fractures should be
screened for the presence of osteoporosis by DEXA.
Management
Management of fracture in the acute stage requiresadequate pain relief, with opiates if necessary, reduction
of the fracture to restore normal anatomy, and immobilization of the affected limb to promote healing. This can be achieved either by the use of an external cast or splint or by internal fixation. Femoral neck fractures present a special management problem since non-union and avascular necrosis are common. This is especially true with intracapsular hip fractures, which should be treated by joint replacement surgery.
Following the fracture, rehabilitation is required with physiotherapy and a supervised exercise programme (this is especially important in older patients to prevent muscle-wasting and loss of mobility).
Generalised musculoskeletal pain
Clinical assessmentClinical history and examination can often indicate the underlying cause . Relentlessly progressive pain occurring in association with weight loss suggests malignant disease with bone metastases. Generalised bone pain may also occur in Paget’s disease if the disease is widespread, but Pagetic pain is usually more focal and
localised to the site of involvement . Widespread pain can occur in OA but this also tends to be localised to sites of involvement, such as the lumbar spine, hips, knees and hands. Signs of OA may be apparent on clinical examination. Osteomalacia can cause generalised bone pain that is associated with bone tenderness and limb girdle weakness.
Fibromyalgia can present with generalised pain particularly affecting the trunk, back and neck. Accompanying features include fatigue, poor concentration and focal areas of hyperalgesia.
Causes of generalised pain
• Metastatic bone disease
• Fibromyalgia
• Joint hypermobility
• Osteomalacia
• Osteoarthritis
• Paget’s disease
• Polymyalgia rheumatica
• Myositis
Investigations
Radionuclide bone scanning is of value in patients suspected of having bone metastases and Paget’s disease. Myeloma should be excluded by plasma and urinary protein electrophoresis. If these results are positive, a radiological skeletal survey should be performed, since the isotope bone scan may be normal in myeloma.Routine biochemistry, vitamin D levels and PTH measurement should be performed if osteomalacia is suspected. In Paget’s disease, ALP may be elevated but can be normal in localised disease. Laboratory investigations are normal in patients with fibromyalgia and benign hypermobility.
Management
Management should be directed towards the underlyingcause. Chronic pain of unknown cause and that associated
with fibromyalgia responds poorly to analgesics
and NSAID, but may respond partially to antineuropathic
agents such as amitriptyline, duloxetine, gabapentin
and pregabalin.
Back pain
A common symptom that affects 60–80% of people at some time in their lives.
Clinical assessment
Mechanical back pain is the most common cause of acute back pain in people aged 20–55,accounts for > 90% of episodes, and is usually acute and associated with heavy lifting , twisting or bending. It is exacerbated by activity and is generally relieved by rest. Usually confined to the lumbar–sacral region, buttock or thigh, is asymmetrical, and does not radiate beyond the knee (which would imply nerve root irritation). On examination, there may be asymmetric local paraspinal muscle spasm and tenderness, and painful restriction of some movements.
After 2 days, 30% are better and 90% have recovered by 6 weeks .Recurrences of pain may occur and about 10–15% go on to develop chronic back pain that may be difficult to treat. Psychological elements, such as job dissatisfaction, depression and anxiety, are important risk factors for the transition to chronic pain and disability. If there is clinical evidence of spinal cord or nerve root compression, or a cauda equina lesion , urgent investigation is needed.
Spinal stenosis presents with leg discomfort on walking that is relieved by rest (pseudoclaudication). Bending forwards or walking uphill may also relieve the pain.
Common causes include Paget, in which enlargement of the vertebrae may encroach on the spinal canal, and OA of the spine, in which osteophytes can have the same effect. Patients may adopt a characteristic simian posture, with a forward stoop and slight flexion at hips and knees.
Degenerative disc disease is a common cause of
chronic low back pain. Prolapse of an intervertebral
disc presents with nerve root pain, which can be accompanied by a sensory deficit, motor weakness, and asymmetrical reflexes. Examination may reveal a positive
sciatic or femoral stretch test. About 70% improve by 4 weeks. Inflammatory back pain due to seronegative spondyloarthritis has a gradual onset and almost always occurs before the age of 40.
It is associated with morning stiffness and improves with movement.
Spondylolisthesis may cause back pain that is typically aggravated by standing and walking. Occasionally,diffuse idiopathic skeletal hyperostosis (DISH )
can cause back pain but it is usually asymptomatic.
Arachnoiditis is a rare cause of chronic severe low
back pain.
It is due to chronic inflammation of the nerve root sheaths in the spinal canal and can complicate meningitis, spinal surgery, or myelography with oil-based contrast agents.
Causes of low back pain
• Mechanical back pain
• Prolapsed intervertebral disc
• Osteoarthritis
• Vertebral fracture
• Spinal stenosis
• Paget’s disease
• Spondylolysis
• Bone metastases
• Spondylolisthesis
• Arachnoiditis
• Scheuermann’s disease
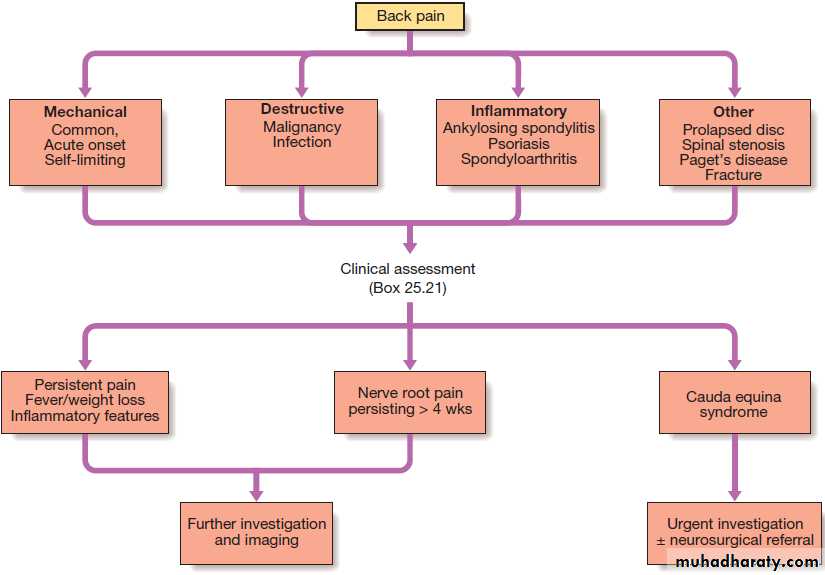
Initial triage assessment of back pain.

Features of mechanical low back pain
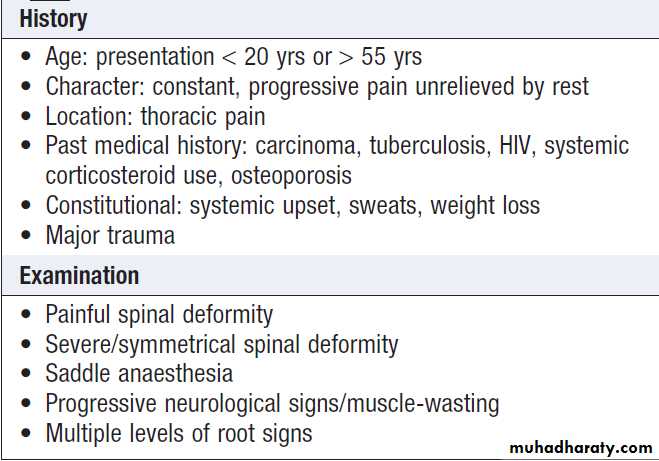
Red flags for possible spinal pathology
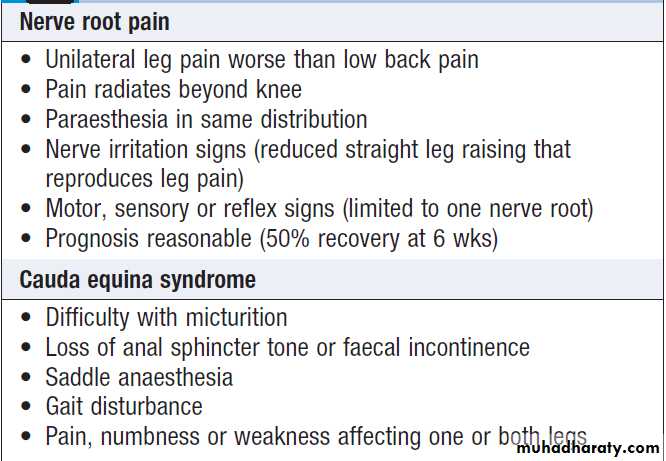
Clinical features of radicular pain
InvestigationsAre not required in acute mechanical back pain. Those with persistent pain (> 6 weeks) or red flags should undergo further investigation. MRI is the investigation of choice since it can demonstrate spinal stenosis, cord or nerve root compression, as well as inflammatory changes and infectious causes such as spinal abscess. Plain radiographs in suspected of having vertebral compression fractures, OA and degenerative disc disease. If metastatic disease is suspected, bone scan or SPECT should be considered. Additional investigations that may be required include ESR and CRP (to screen for sepsis and inflammatory disease), protein and urinary electrophoresis (for myeloma) and prostate specific antigen (for prostate carcinoma).
Management
Education is important in patients with mechanical backpain. It should emphasise the self-limiting nature of the
condition and the fact that exercise is helpful rather than
damaging. Regular analgesia and/or NSAIDs may be
required to improve mobility and facilitate exercise.
Return to work and normal activity should take place as
soon as possible. Bed rest is not helpful and may increase
the risk of chronic disability.
Referral for physiotherapy or manipulation should be considered if a return to normal activities has not been achieved by 6 weeks. Low-dose tricyclic antidepressant drugs may help pain, sleep and mood.
Other treatment modalities that are occasionally used
include epidural and facet joint injection, traction and
lumbar supports, though there is little evidence to
support their use. Malignant disease, osteoporosis,
Paget’s disease and spondyloarthropathies
require specific treatment of the underlying condition.
Surgery is required in less than 1% of patients with
low back pain but may be needed in spinal stenosis, in
spinal cord compression and in some patients with
nerve root compression.

Management of low back pain
Neck painNeck pain is a common symptom that can occur following
injury (for example, whiplash), after falling asleep in
an awkward position, as a result of stress, or in association
with OA of the spine.
Most cases resolve spontaneously or with a short
course of NSAID or analgesics, and a soft collar. Patients
with persistent pain that follows a nerve root distribution
and those with neurological signs and symptoms
should be investigated by MRI scan, and if necessary
referred for a neurosurgical opinion.
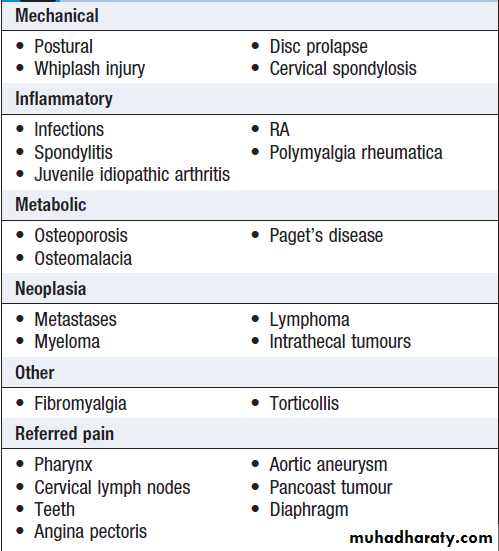
Causes of neck pain
Shoulder painShoulder pain is a common complaint in both genders
> 40, and is most often due to degenerative disease of tendons in the rotator cuff .
Management is symptomatic, with analgesics, NSAID, local corticosteroid injections and physiotherapy aimed at restoring normal movement and function. Surgery may be required in debilitating symptoms in association with rotator cuff tears. Adhesive capsulitis (frozen shoulder) presents with upper arm pain that can progress over 4–10 weeks before subsiding over a similar time course. Restriction of glenohumeral movement is characteristic. In the early phase, there is marked anterior joint/ capsular tenderness and stress pain in a capsular pattern; later there is painless restriction, of movements.
Frozen shoulder is more common in diabetes mellitus,
but may also be triggered by a rotator cuff tear, localtrauma, myocardial infarction or hemiplegia.
Treatment in the early stage is with analgesia, intra- and extracapsular steroid injection, and regular ‘pendulum’ exercises of the arm to prevent the capsule from over-tightening.
Mobilising and strengthening exercises are the sole
treatment in the painless ‘frozen’ stage. The natural history is for slow but complete recovery, sometimes
taking up to 2 years.
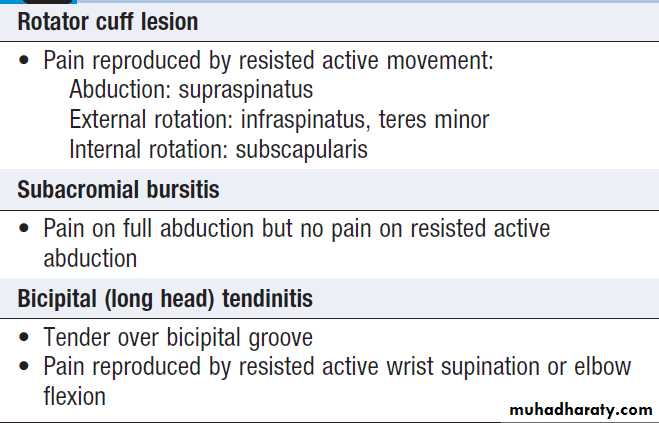
Clinical findings in shoulder pain
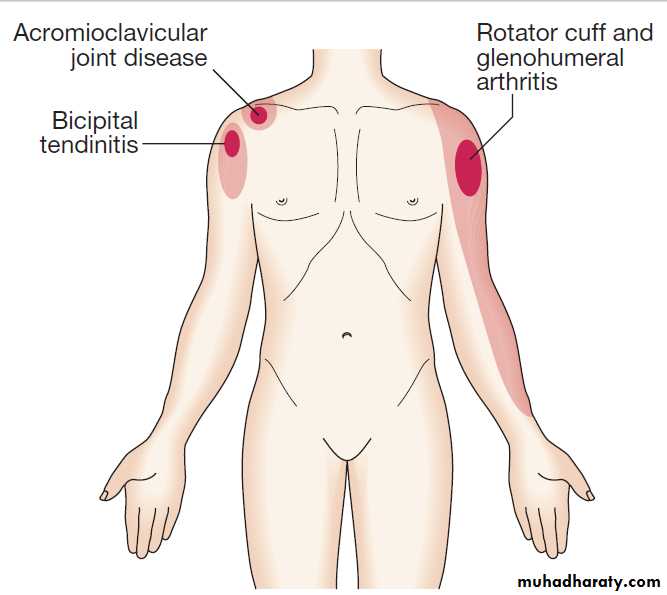
Pain patterns around the shoulder. The dark shading indicates sites of maximum pain.
Elbow painThe most common causes are repetitive strain injury
affecting the lateral epicondyle (tennis elbow) and
medial epicondyle (golfer’s elbow) .
Management
is by rest, analgesics and topical or systemic
NSAID. Symptoms may also respond to local application
of glyceryl trinitrate patches. Local corticosteroid
injections may be required in resistant cases. Olecranon
bursitis can also follow local repetitive trauma but other
causes include infections, gout and RA.
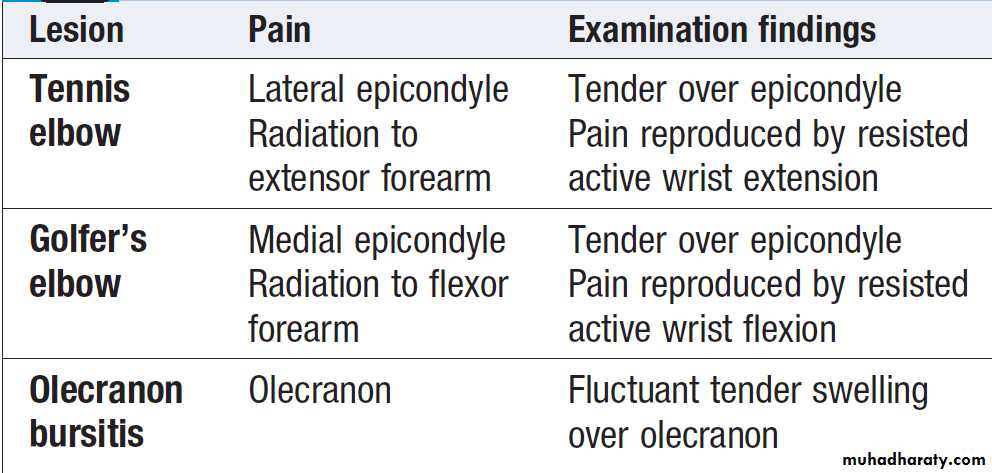
Local causes of elbow pain
Hand and wrist pain
Pain from hand or wrist joints is well localised to the affected joint, except for pain from the first metacarpal joint, commonly targeted by OA; although maximal at the thumb base, the pain often radiates down the thumb and to the radial aspect of the wrist.Non-articular causes of hand pain include:
• Tenosynovitis: flexor or extensor (pain and swelling, with or without fine crepitus on volar or extensor aspect).
De Quervain’s tenosynovitis involves the tendon sheaths of abductor pollicis longus and extensor pollicis brevis, and produces pain maximal over the radial aspect of the distal forearm and wrist. It usually occurs as the result of a repetitive strain injury.
There is tenderness (with or without warmth, linear swelling and fine crepitus) over the distal radius and marked pain on forced ulnar deviation of the wrist with the thumb held across the patient’s palm (Finkelstein’s sign).
• Raynaud’s phenomenon .
• C8/T1 radiculopathy.
• Reflex sympathetic dystrophy .
Hip pain
Pain from the hip joint is usually maximal deep in the anterior groin, with variable radiation to the buttock, anterolateral thigh, knee or shin . Trochanteric bursitis is a common cause , typically affecting obese women, and occurring in isolation or secondary to an abnormal gait, such as in hip or knee OA. Pain in the hip region may also be referred from the back. Root entrapment can cause pain in the lateral thigh (T12–L1) or the inguinal region and lateral thigh (L2–4), but is worsened by coughing and straining more than by movement and is often accompanied by sensory disturbance.Other less common causes include psoas abscess, retroperitoneal haemorrhage or pelvic inflammation, which can cause inguinal and lateral thigh pain that is aggravated by resisted hip flexion.
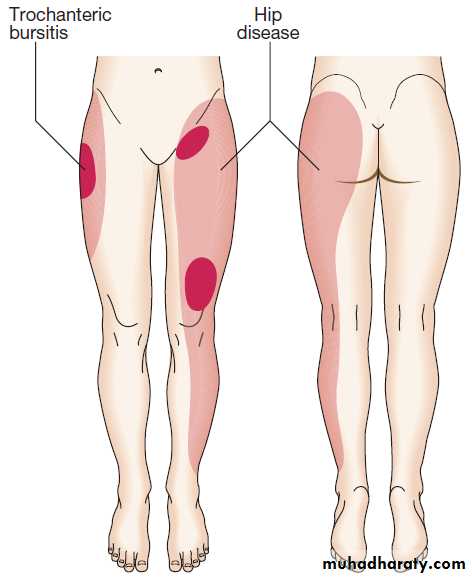
Pain patterns of hip disease and trochanteric bursitis.
The dark shading indicates sites of maximum pain.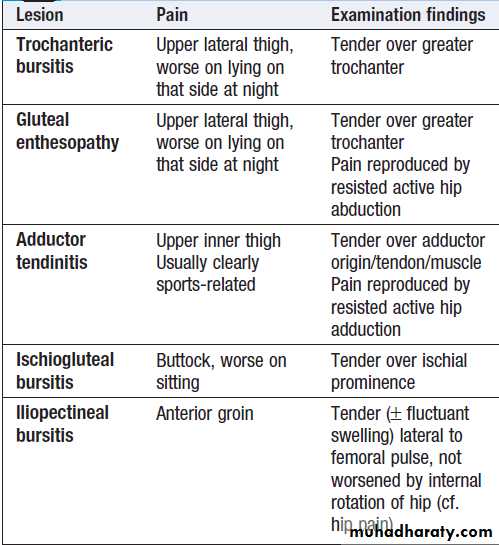
Local causes of hip pain
Knee painThe most common cause of knee pain is OA. Pain that is associated with locking of the knee (sudden painful inability to extend fully) is usually due to a meniscal tear or osteochondritis dissecans. Referred pain from the hip may present at the knee and is reproduced by hip not knee movement. Pain from periarticular lesions is well localised to the involved structure . Anterior knee pain may be due to bursitis occurring as the result of repetitive occupational kneeling, as well as infection and gout. Rarely, anterior knee pain may be the result of chondromalacia patellae, in which degenerative changes of the articular cartilage occur.
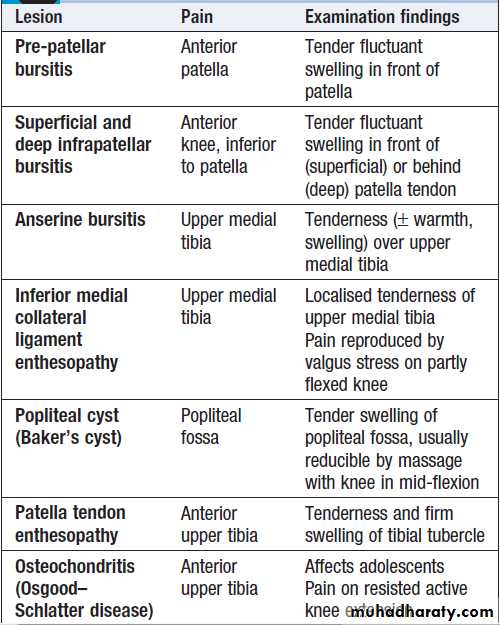
Local causes of knee pain
Ankle and foot painPain from the mortice joint of the ankle (the tibiofibular–
talar joint) is felt between the malleoli and is worse on
weight-bearing. Pain from the subtalar joint is also worse
on weight-bearing on uneven surfaces. The mortice joint
is commonly affected by OA, whereas RA tends to affect
the subtalar joint. Pain under the heel can arise from
plantar fasciitis or subcalcaneal bursitis. Pain affecting
the back of the heel may be due to Achilles tendinitis or
bursitis. Patients with seronegative spondyloarthritis
may develop enthesopathy affecting this region, resulting
in plantar fasciitis, which presents with pain and tenderness under the heel, or as Achilles enthesitis, which presents with pain at the tendon insertion into the calcaneus.
The MTP joints of the feet are commonly involved in RA. The presentation is with pain on walking below the metatarsal heads, often described as ‘walking
on marbles’. Patients with active inflammation of the
MTP joints have pain when the forefoot is squeezed .
Involvement of the first MTP joint is common in OA and is associated with a valgus deformity (hallux valgus). This joint is also a classical target in acute gout.
Claw foot (pes cavus) can be associated with anterior foot pain and is characterised by a high arch and clawing of the toes. It may be an isolated phenomenon or secondary to neurological disorders such as Friedreich’s ataxia
or spina bifida .
Management
Bursitis and enthesitis resistant to standard measures mayrespond to local steroid injections. Morton’s neuroma is
the name given to an entrapment neuropathy of the
interdigital nerves of the feet, which presents with shooting
pain that is usually located between the third and
fourth metatarsal heads. Women are most commonly
affected. Local sensory loss and a palpable tender swelling between the metatarsal heads may be detected. Footwear adjustment, with or without a local corticosteroid
injection, often helps but surgical decompression may be
required if symptoms persist.
Muscle pain and weakness
Muscle pain and weakness can arise from a variety ofcauses. It is important to distinguish between a subjective
feeling of generalised weakness or fatigue, and an
objective weakness with loss of muscle power and function.
Clinical assessment
Proximal muscle weakness suggests the presence of a
myopathy or myositis, which typically causes difficulty
with standing from a seated position, squatting and
lifting overhead. Worsening of symptoms on exercise and post-exertional cramps suggest a metabolic myopathy, such as glycogen storage disease . A strong family history and onset in childhood or early adulthood suggests muscular dystrophy .
Alcohol excess can cause an inflammatory myositis and atrophy of type 2 muscle fibres.
Proximal myopathy may be a complication of corticosteroid therapy and of osteomalacia. Myopathy and myositis can also occur in association with statin use and viral infections, including HIV infection, when it may be due to HIV itself or treatment with zidovudine.
Clinical examination should document the presence, pattern and severity of muscle weakness, and the latter should be assessed using the Medical Research Council (MRC) scale, in which muscle strength is graded on a six-point scale ranging from no power (0) to full power (5).
Investigations
Investigations should include routine biochemistry andhaematology. ESR and CRP, may be raised in inflammatory myositis and CK. Serum 25(OH) vitamin D levels and PTH should be checked in suspected osteomalacia. Raised CK levels suggest muscle pathology but do not establish the cause. Muscle biopsy and electromyography (EMG) are usually required to make the diagnosis, but MRI can be used to identify focal areas of muscle abnormality and increase the diagnostic yield from muscle biopsies.
Management
Management is determined by the cause but all patients
with muscle disease may benefit from physiotherapy
and graded exercises to maximise muscle function.
Inflammatory
• Polymyositis • Dermatomyositis • Inclusion body myositis • Polymyalgia rheumaticaEndocrine
• Hypothyroidism • Hyperthyroidism • Cushing’s syndrome • Addison’s disease
Metabolic
• Myophosphorylase deficiency • Phosphofructokinase deficiency • Hypokalaemia • Carnitine deficiency • Myoadenylate deaminase deficiency • Osteomalacia
Drugs/toxins
• Alcohol • Cocaine • Fibrates • Statins • Penicillamine • Zidovudine
Infections
• Viral (HIV, CMV, rubella, Epstein–Barr, echo) • Parasitic (schistosomiasis, cysticercosis, toxoplasmosis)
• Bacterial (Clostridium perfringens, staphylococci, TB , Mycoplasma)
Causes of proximal muscle pain or weakness
PRINCIPLES OF MANAGEMENT OF MUSCULOSKELETAL DISORDERS
Although management of musculoskeletal disease
depends on the underlying diagnosis, certain aspects of
management are common to many disorders. The
general aims of management are to:
• educate the patient
• control pain
• optimise function
• modify the disease process where this is possible
• identify and treat related comorbidity.
The management plan should be individualised and patient-centred, should involve all necessary members of the multidisciplinary team.
It must also take into account:
• the person’s daily activity requirements, and work
and recreational aspirations
• risk factors and associations of the musculoskeletal
condition (obesity, muscle weakness, nonrestorative sleep)
• the person’s perceptions and knowledge of the
condition
• medications and strategies already tried by the patient
• comorbid disease and its therapy
• the availability, costs and logistics of appropriate
evidence-based interventions.
Simple and safe interventions should be tried first.
Symptoms and signs will change with time, so the plan
requires regular review and re-adjustment. Effective
management may require the expertise of a variety of
health professionals, with a coordinated multidisciplinary
team approach.
Core interventions that should be considered for
everyone with a painful musculoskeletal condition
are listed in Box .
There are also other nonpharmacological and drug options, the choice depending largely on the nature and severity of the diagnosis.
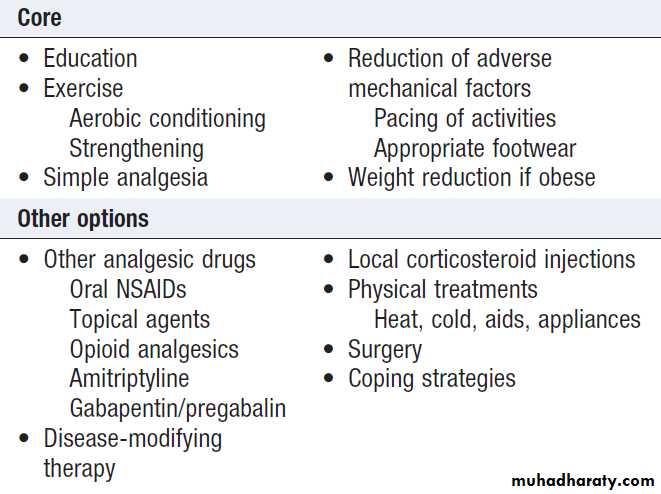
Core interventions for patients with
rheumatic diseasesEducation and lifestyle interventions
EducationPatients must always be informed about the nature of
their condition and its investigation, treatment and
prognosis, as education can improve outcome. Information
and therapist contact can reduce pain and disability,
improve self-efficacy and reduce the health-care costs . Education can be provided through one-to-one
discussion, written literature, patient-led group education
classes and interactive computer programs. Inclusion of the patient’s partner or carer is often appropriate.
Exercise
• Aerobic fitness training can produce long-term reduction in pain and disability. It improves well-being, encourages restorative sleep and benefits common comorbidity such as obesity, diabetes, chronic heart failure and hypertension.
• Local strengthening exercise for muscles that act
over compromised joints also reduces pain and disability, with improvements in the reduced muscle strength, proprioception, coordination and balance that associate with chronic arthritis.

Education and exercise in the management of arthritis
Joint protectionExcessive impact-loading and adverse repetitive use of
a compromised joint or periarticular tissue can often be
reduced: for example, cessation of contact sports, or
altered use of machinery or tools at the workplace.
Simple ‘pacing’ of activities – dividing physically
onerous tasks into shorter segments with brief breaks in
between – is helpful. Use of shock-absorbing footwear
with thick soft soles can reduce impact-loading through
feet, knees, hips and back, and improve symptoms at
these sites. A walking stick held on the contralateral side
takes the weight off a painful hip, knee or foot.
Weight loss
Obesity aggravates pain at most sites of the body
through increased mechanical strain and is a risk factor
for more rapid progression of joint damage in patients
with arthritis. This should be explained to obese patients and strategies offered on how to lose and then maintain an appropriate weight .
Pharmacological treatment
Analgesics
Paracetamol (1 g up to 4 times daily) is the oral analgesic
of first choice and, if successful, the preferred long-term
oral analgesic. It inhibits prostaglandin synthesis in the
brain but has less effect on peripheral prostaglandin
production. It is generally well tolerated and has few
adverse effects and drug interactions.
There is a possible increased risk of both GI events and CVD with chronic usage, but it is uncertain whether this is due to the underlying disease or the drug itself.
If paracetamol fails, it can be used in combination with opioids such as codeine and dihydrocodeine in compound analgesic preparations like co-codamol (codeine and paracetamol) or co-dydramol (dihydrocodeine and paracetamol).
Although these are more effective than paracetamol, side-effects include constipation, headache and confusion, especially in the elderly. The centrally acting analgesics tramadol and meptazinol may be useful for temporary control of severe pain unresponsive to other measures. Both may cause nausea, bowel upset, dizziness and somnolence, and withdrawal symptoms after chronic use.
The non-opioid analgesic nefopam (30–90 mg 3 times daily) can help moderate pain, though side-effects (nausea, anxiety, dry mouth) often limit its use. Patients with severe or intractable pain may require stronger opioid analgesics such as oxycodone and morphine.
Non-steroidal anti-inflammatory drugs
These are among the most widely prescribed drugs, but their use has declined over recent years because of concerns about an increased risk of CVD. Oral NSAIDs are particularly useful in the management of pain that has an inflammatory component, and a long-acting NSAID taken in the evening may help reduce early morning stiffness. There is marked variability in individual tolerance and response; patients who do not respond to one NSAID may still gain relief from another. The mechanism of action is through inhibition of prostaglandin H synthase and cyclo-oxygenase (COX) enzymes.
Arachidonic acid,derived from membrane phospholipids, is metabolized to produce prostaglandins and leukotrienes by the COX and 5-lipoxygenase pathways respectively .
There are two isoforms of COX, encoded by different
genes. COX-1 is expressed and fulfils a ‘housekeeping’ function in the gastric mucosa, platelets and kidneys. The COX-2 enzyme is largely induced at sites of inflammation, producing prostaglandins that cause local pain and inflammation, but COX-2 is also up-regulated in the CNS, where it plays a role in the central mediation of pain and fever. Traditional NSAIDs, such as ibuprofen, diclofenac and naproxen, inhibit both COX enzymes, whereas newer celecoxib and etoricoxib, selectively inhibit COX-2.
Whilst NSAIDs have anti-inflammatory activity,
they are not thought to have a disease-modifying effectin either OA or inflammatory rheumatic diseases.
Non-selective NSAIDs can damage the gastric and
duodenal mucosal barrier and are associated with
an increased risk of upper GI ulceration, bleeding and perforation.
Dyspepsia is a poor guide to the presence of NSAID-associated ulceration and bleeding .
Co-prescription of omeprazole (20 mg daily) or misoprostol (200 μg twice or 3 times daily) reduces but does not eliminate NSAID-induced ulceration and bleeding, but H2-antagonists in standard doses are ineffective.
The selective COX-2 inhibiter are much less likely to cause gastrointestinal toxicity but benefit is attenuated in patients on low dose aspirin. In the UK, NICE guidelines advise that PPI should be co-prescribed with all NSAIDs, including COX-2 selective NSAIDs, even though the risk of GI events with these is low. Since chronic PPI therapy is associated with an increased risk of hip fracture, the merits of giving PPI therapy with a COX-2 selective drug need to be weighed up carefully. Other SE include fluid retention and renal impairment due to inhibition of renal prostaglandin production, non-ulcer-associated dyspepsia, abdominal pain and altered bowel habit, and rashes. Interstitial nephritis, asthma and anaphylaxis can also occur but are rare. Because of the risk of adverse effects, NSAIDs should be used with great care in the elderly .
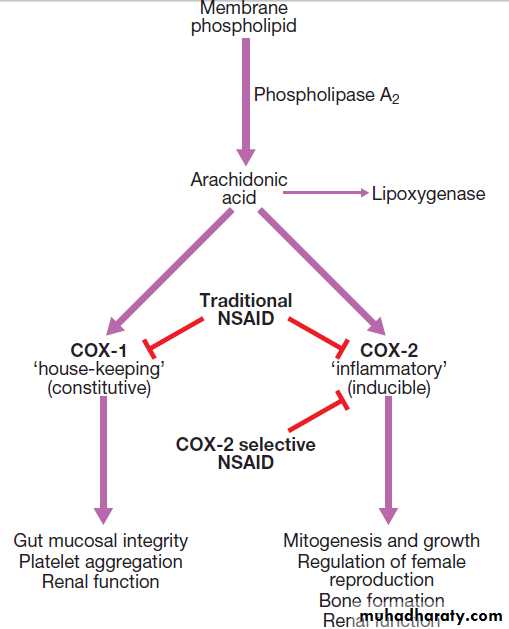
COX-1 and COX-2 pathways.
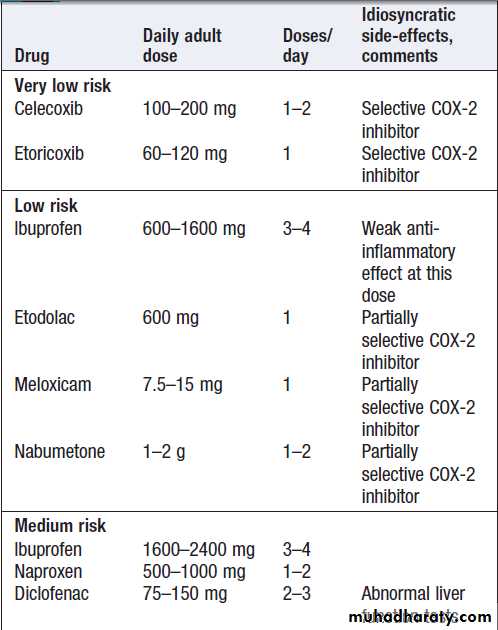
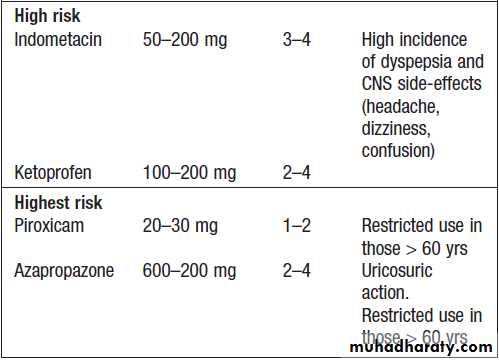
Commonly used NSAIDs and their relative
risk of gastrointestinal bleeding and perforationRisk factors for NSAID-induced ulcers
• Age > 60 yrs*• Past history of peptic ulcer*
• Past history of adverse event with NSAID
• Concomitant corticosteroid use
• High-dose or multiple NSAID
• High-risk NSAID
*The most important risk factors.

Use of oral NSAID in old age
Recommendations for the use of NSAID• Use with caution in patients with cardiovascular disease
• Use the lowest dose for the shortest time possible to control symptoms
• Avoid NSAID in patients on warfarin
• Allow 2–3 weeks to assess efficacy. If response is
inadequate, consider trial of another NSAID
• Never prescribe more than one NSAID at a time
• Co-prescribe proton pump inhibitor for patients with risk
factors for gastrointestinal adverse effects
Topical agents
Topical NSAID creams and gels and capsaicin cream can help in the treatment of OA and superficial periarticular lesions affecting hands, elbows and knees. They may be used as monotherapy or as an adjunct to oral analgesics. Topical NSAIDs can penetrate superficial tissues and even reach the joint capsule. Initial application causes a burning sensation but continued use depletes presynaptic substance P, with subsequent pain reduction that is optimal after 1–2 weeks.
Non-pharmacological interventions
Physical and occupational therapyLocal heat, ice packs, wax baths and other local external
applications can induce muscle relaxation and temporary
relief of symptoms in a range of rheumatic diseases.
Hydrotherapy induces muscle relaxation and facilitates
Enhanced movement in a warm, pain-relieving
environment without the restraints of gravity and
normal load-bearing. Various manipulative techniques
may also help improve restricted movement. Splints can give temporary rest and support for painful joints and periarticular tissues, and prevent harmful involuntary postures during sleep.
Prolonged rest, however, must be avoided.
Orthoses are more permanent appliances used to
reduce instability and excessive abnormal movement.They include working wrist splints, knee orthoses, and
iron and T-straps to control ankle instability. Aids and appliances can provide dignity and independence to patients with respect to activities of daily living. Common examples are a raised toilet seat, raised chair height, extended handles on taps, a shower instead of a bath, thick-handled cutlery, and extended ‘hands’ to pull on tights and socks. Full assessment and advice from an occupational therapist maximise the benefits of these.
Surgery
A variety of surgical interventions can relieve pain and conserve or restore function in patients with bone, joint and periarticular disease .Soft tissue release ,tenosynovectomy may reduce inflammatory symptoms, improve function.Synovectomy of joints does not prevent disease progression but may be indicated for pain relief when drugs, physical therapy and intra-articular injections have provided insufficient relief. The main approaches for damaged joints are osteotomy (cutting bone to alter joint mechanics and load transmission), excision arthroplasty (removing part or all of the joint), joint replacement and arthrodesis (joint fusion). Surgical fixation of fractures.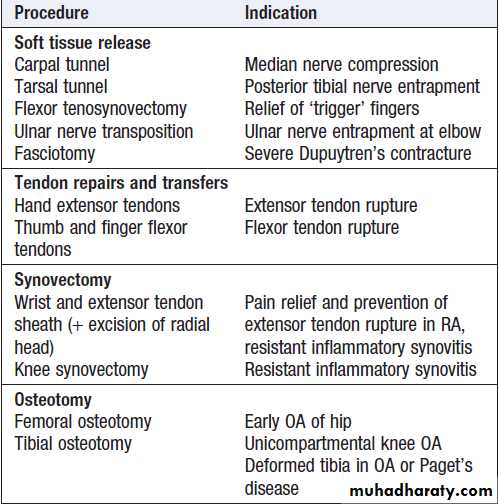
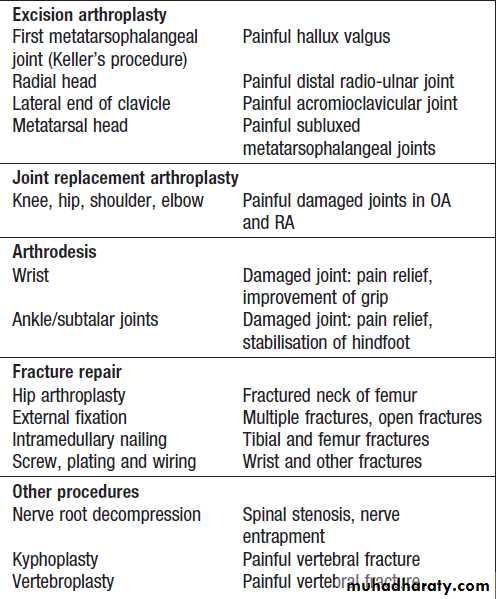

Surgical procedures in rheumatology and
bone diseaseSelf-help and coping strategies
These help patients to cope better with, and adjust to,chronic pain and disability. They may be useful at any stage but are particularly so for patients with incurable
problems, who have tried all available treatment options.
The aim is to increase self-management through self-assessment and problem-solving, so that patients can
recognise negative but potentially remediable aspects of
their mood (stress, frustration, anger or low self-esteem)
and their situation (physical, social, financial). Involvement of the spouse or partner in mutual goal setting can improve partnership adjustment. Such approaches are often an element of group education and pain clinics, but may require more formal clinical psychological input.

Self-help and coping strategies
OSTEOARTHRITIS (OA)
OA is by far the most common form of arthritis. It is strongly associated with ageing and is a major cause of pain and disability in older people. Osteoarthritis is characterised by focal loss of articular cartilage, subchondral osteosclerosis, osteophyte formation at the joint margin, and remodelling of joint contour with enlargement of affected joints. Inflammation can occur but is not a prominent feature.Joint involvement in OA follows a characteristic distribution, mainly targeting the hips, knees, PIP and DIP joints of the hands, neck and lumbar spine .
The prevalence of OA rises progressively with age. Symptoms attributable to OA are more prevalent in women, except at the hip, men are equally affected.
Pathophysiology
OA is a complex disorder with both genetic and environmental components . Repetitive adverse loading of joints during occupation or competitive sports is also an important predisposing factor in farmers (hip OA), miners (knee OA) and elite or professional athletes (knee OA). Congenital abnormalities, such as slipped femoral epiphysis, are also associated with a high risk, and this is also thought to be the explanation for the increased risk of OA in Paget’s disease of bone.
Obesity is another strong risk factor. Although this
is likely to be due in part to increased mechanical loadingof the joints, it has been speculated that cytokines
released from adipose tissue may also play a role. The
increased incidence of OA in women has led to speculation
that sex hormones may play a causal role.
Some studies have shown a lower prevalence of OA in women who use hormone replacement therapy (HRT), as compared with non-users.
Genetic factors play a key role in the pathogenesis of OA, and family-based studies have estimated that the heritability of OA ranges between about 43% at the knee to between 60% and 65% at the hip and hand, respectively.
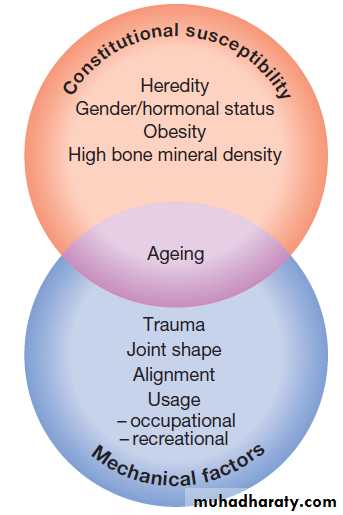
Risk factors for the development of osteoarthritis.
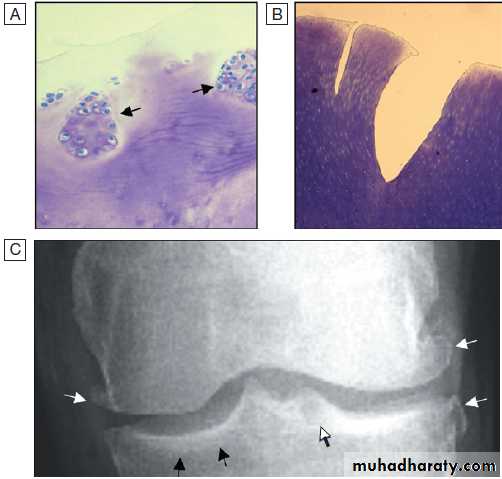
Pathological changes in osteoarthritis.
A Abnormalnests of proliferating chondrocytes (arrows) interspersed with matrix devoid
of normal chondrocytes.
B Fibrillation of cartilage in OA.
C Radiograph
of knee joint affected by OA, showing osteophytes at joint margin (white arrows), subchondral sclerosis (black arrows) and subchondral cyst (open arrow).
Clinical features
The main presenting symptoms are pain and functionalrestriction in a patient over the age of 45, but more
often over 60 years. The causes of pain in OA are not
completely understood but may relate to increased
pressure in subchondral bone (mainly causing night
pain), trabecular microfractures, capsular distension and
low-grade synovitis, or may result from bursitis and
enthesopathy secondary to altered joint mechanics.
Radiological evidence of OA is very common in
middle-aged and older people, and may coexist with other conditions, so it is important to remember that pain in a patient with OA may be due to another cause.
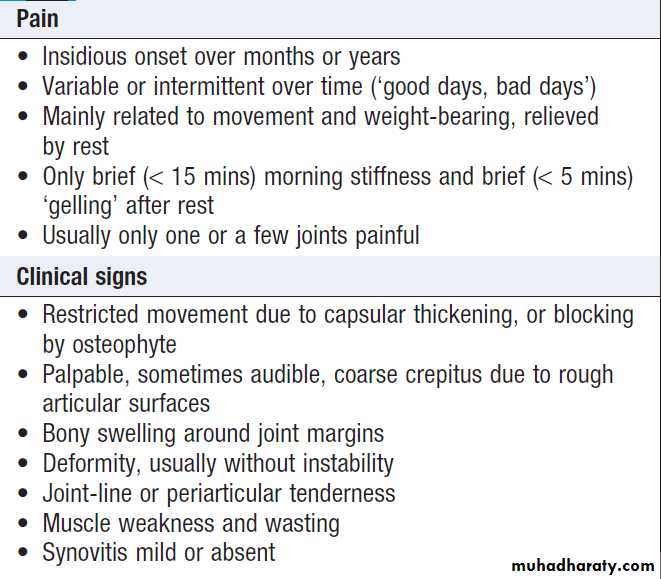
Symptoms and signs of osteoarthritis
Generalised nodal OASome patients are asymptomatic whereas others develop pain, stiffness and swelling of one or more PIP joints of the hands from the age of about 40 years onward. Gradually, these develop posterolateral swellings on each side of the extensor tendon that slowly enlarge and harden to become Heberden’s (DIP) and Bouchard’s (PIP) nodes. Typically, each joint goes through a phase of episodic symptoms (1–5 years) while the node evolves and OA develops. Once OA is fully established, symptoms may subside and hand function often remains good. Affected joints are enlarged as the result of osteophyte formation and often show characteristic lateral deviation, reflecting the asymmetric focal cartilage loss of OA .
Clinically, it may be detected by the presence of crepitus on joint movement, and squaring of the thumb base.
Generalised nodal OA has a very strong genetic component: the daughter of an affected mother has a 1 in 3 chance of developing nodal OA herself. People with
nodal OA are at increased risk of OA at other sites,
especially the knee.

Characteristics of generalised nodal OA
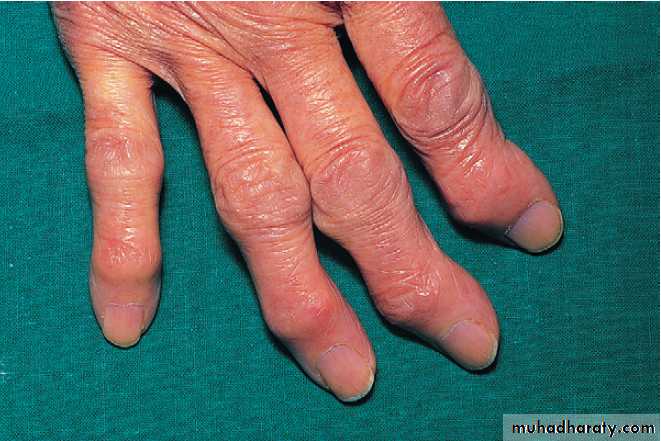
Nodal osteoarthritis. Heberden’s nodes and lateral (radial/ ulnar) deviation of distal interphalangeal joints, with mild Bouchard’s nodes at the proximal IP joints.
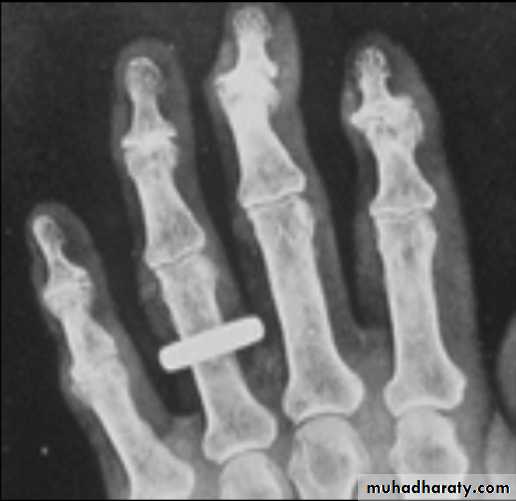
X-ray appearances in hand osteoarthritis. There is
marked loss of joint space at all of the distal interphalangeal joints, with osteophyte formation most marked at the first and second DIP joints. The
fifth proximal interphalangeal joint also shows loss of joint space with osteophyte formation.
Knee OA
OA principally targets the patello-femoral and medialtibio-femoral compartments at this site but eventually
spreads to affect the whole of the joint (Fig.). It may
be isolated or occur as part of generalised nodal OA.
Most patients, particularly women, have bilateral and
symmetrical involvement. With men, trauma is a more
important risk factor and may result in unilateral OA.
The pain is usually localised to the anterior or medial
aspect of the knee and upper tibia. Patello-femoral pain
is usually worse going up and down stairs or inclines.
Posterior knee pain suggests the presence of a complicating popliteal cyst (Baker’s cyst). Prolonged walking, rising from a chair, getting in or out of a car, or bending to put on shoes and socks may be difficult. Local examination findings may include:
• a jerky, asymmetric (antalgic) gait with less time
weight-bearing on the painful side
• a varus less commonly valgus, and/or fixed flexion deformity
• joint-line and/or periarticular tenderness
(secondary anserine bursitis and medial ligament
enthesopathy causing tenderness of the upper medial tibia)
• weakness and wasting of the quadriceps muscle
• restricted flexion/extension with coarse crepitus
• bony swelling around the joint line.
Calcium pyrophosphate dihydrate (CPPD) crystal
deposition in association with OA is most common
at the knee.
This may result in a more overt inflammatory component (stiffness, effusions) and super-added acute attacks of synovitis (‘pseudogout’), which may predict more rapid radiographic and clinical progression.
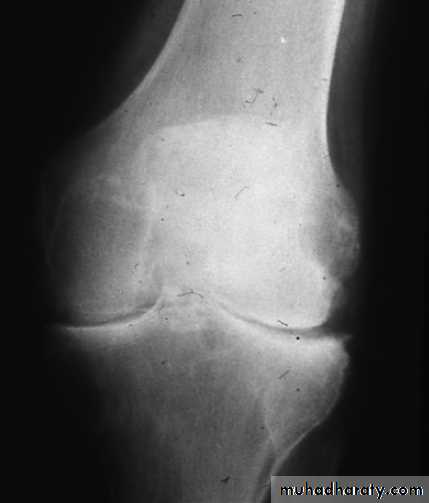
X-ray appearances in knee osteoarthritis. There is almost complete loss of joint space affecting both compartments,
And sclerosis of subchondral bone.
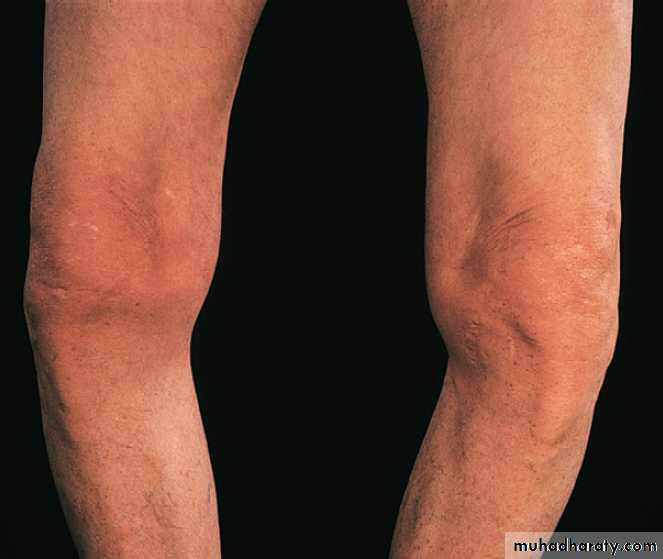
Typical varus deformity resulting from marked medial
tibio-femoral osteoarthritis.Hip OA
Hip OA most commonly targets the superior aspect ofthe joint . This is often unilateral at presentation,
frequently progresses with superolateral migration
of the femoral head, and has a poor prognosis. The less
common central (medial) OA shows more central cartilage
loss and is largely confined to women.
It is often bilateral at presentation and may associate with generalised nodal OA.
It has a better prognosis than superior hip OA and progression to axial migration of the femoral
head is uncommon.
The hip shows the best correlation between symptoms
and radiographic change. Hip pain is usually
maximal deep in the anterior groin, with variable radiation to the buttock, anterolateral thigh, knee or shin.
Lateral hip pain, worse on lying on that side with tenderness over the greater trochanter, suggests secondary trochanteric bursitis.
Common functional difficulties are the same as for knee OA; in addition, restricted hip abduction in women may cause pain on intercourse.
Examination may reveal:
• an antalgic gait• weakness and wasting of quadriceps and gluteal muscle
• pain and restriction of internal rotation with the hip
flexed – the earliest and most sensitive sign of hip
OA; other movements may subsequently be restricted and painful
• anterior groin tenderness just lateral to the femoral
pulse
• fixed flexion, external rotation deformity of the hip
• ipsilateral leg shortening with severe joint attrition
and superior femoral migration.
Although obesity is not a major risk factor for development
of hip OA, it is associated with more rapid progression.
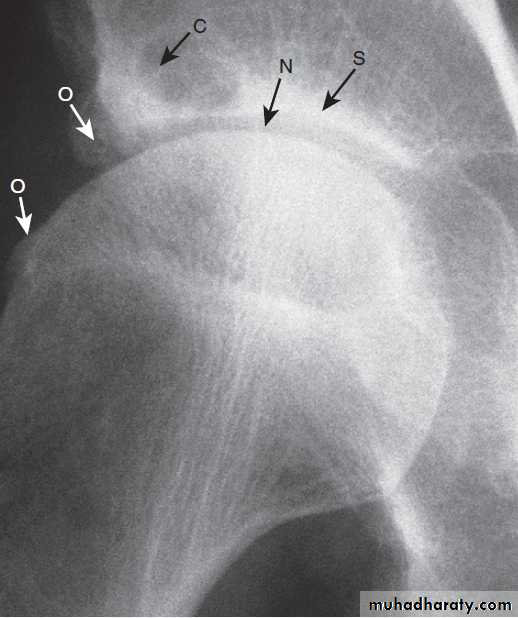
X-ray of hip showing changes of osteoarthritis. Note
the superior joint space narrowing (N), subchondral sclerosis (S), marginalosteophytes (white arrows) and cysts (C).
Spine OA
The cervical and lumbar spine are predominantly targeted, then referred to as cervical spondylosis and lumbar spondylosis, respectively . Spine OA may occur in isolation or as part of generalised OA. The typical presentation is with pain localised to the low back region or the neck, although radiation of pain to the arms, buttocks and legs may also occur due to nerve root compression. The pain is typically relieved by restand worse on movement. On physical examination, the
range of movement may be limited and loss of lumbar
lordosis is typical. The straight leg-raising test or femoral
stretch test may be positive and neurological signs may
be seen in the legs where there is complicating spinal
stenosis or nerve root compression.
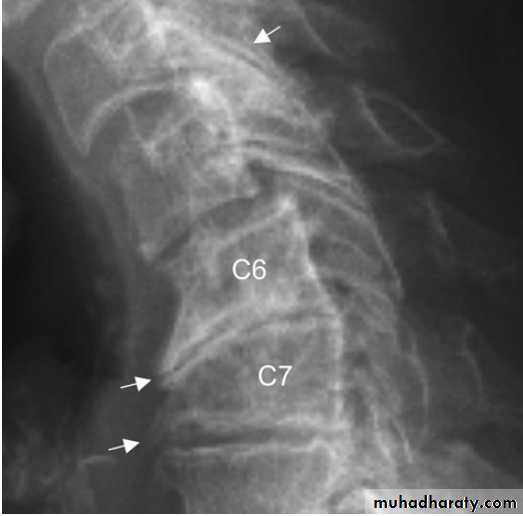
X-ray of spine showing typical changes of
osteoarthritis. Cervical spondylosis showing disc space narrowingbetween C6 and C7, osteophytes at the anterior vertebral body margins (arrows) and osteosclerosis at the apophyseal joints.
Early-onset OA
Unusually, typical symptoms and signs of OA may
present before the age of 45. In most cases, a single joint
is affected and there is a clear history of previous trauma.
However, specific causes of OA need to be considered
in people with early-onset disease affecting several
joints, especially those not normally targeted by OA,
rare causes need to be considered . Kashin–
Beck disease is a rare form of OA that occurs in children,
typically between the ages of 7 and 13, in some regions of China. The cause is unknown but suggested predisposing
factors are selenium deficiency and contamination
of cereals with mycotoxin-producing fungi.
Causes of early-onset osteoarthritis
Monoarticular• Previous trauma, localised instability
Pauciarticular or polyarticular
• Juvenile idiopathic arthritis
• Metabolic or endocrine disease Haemochromatosis ,
Ochronosis , Acromegaly
• Spondylo-epiphyseal dysplasia
• Late avascular necrosis
• Neuropathic joint
• Kashin–Beck disease
Erosive OA
This term is used to describe rare patients with hand OA
who have a more prolonged symptom phase, more overt
inflammation, more disability and worse outcome than
those with nodal OA. Distinguishing features include
preferential targeting of PIP joints, subchondral erosions
on X-rays, occasional ankylosis of affected joints and
lack of association with OA elsewhere.
It is unclear whether erosive OA is part of the spectrum of hand OA or a discrete subset.
Investigations
A plain X-ray will show one or more of the typical featuresof OA . In addition to providing diagnostic information, X-rays are used to assess the severity of structural change, which is useful if joint replacement surgery is being considered. Non-weight-bearing postero-anterior views of the pelvis are adequate for assessing hip OA. Patients with suspected knee OA should have standing antero-posterior radiographs taken to assess tibio-femoral cartilage loss, and a flexed skyline view to assess patello-femoral involvement. Spine OA can often be diagnosed on plain X-ray, which typically shows evidence of disc space narrowing
and osteophytes. If nerve root compression or spinal
stenosis is suspected, MRI should be performed.
Routine biochemistry, haematology and autoantibody
tests are usually normal. Synovial fluid aspiratedfrom an affected joint is viscous with a low cell count.
Radioisotope bone scans performed for other reasons
often show, as an incidental finding, discrete increased
uptake in OA joints due to increased bone remodelling.
Unexplained early-onset OA requires additional
investigation, guided by the suspected underlying condition.
X-rays may show typical features of dysplasia or
avascular necrosis, widening of joint spaces in acromegaly, multiple cysts and chondrocalcinosis in haemochromatosis or
disorganised architecture in neuropathic joints.

Osteoarthritis in old age

Management of osteoarthritis
Education and other general measuresIt is important to explain the nature of the condition
fully, outlining the role of relevant risk factors (obesity,
heredity, trauma) and the fact that established structural
changes are permanent but that pain and function
can improve. The prognosis should also be discussed
(good for nodal hand OA, more optimistic for knee
than hip OA), as should how appropriate action can
improve the prognosis of large-joint OA.
Exercise has beneficial effects in OA, including both strengthening and aerobic exercise, preferably with reinforcement by a physiotherapist .
Quadriceps strengthening exercises are particularly beneficial in knee OA. Weight loss can have a substantial beneficial effect on symptoms if the patient is obese and is probably one of the most effective treatments for reducing pain, particularly in OA of the lower limbs. Shock-absorbing footwear, pacing of activities, use of a walking stick for painful knee or hip OA, or provision of built-up shoes to equalise leg lengths can all improve symptoms.
Analgesics and anti-inflammatory drugs
If symptoms do not respond to non-pharmacologicalmeasures, paracetamol should be given. Addition of a
topical NSAID, and then capsaicin, for knee and hand
OA can also be helpful. Oral NSAID should be considered
in patients who remain symptomatic. These drugs
are significantly more effective than paracetamol and
can be successfully combined with paracetamol or compound analgesics such as co-codamol if the pain is
severe.
Opiates may occasionally be required. For temporary benefit of moderate to severe pain, intra-articular injection of corticosteroids can be helpful (see below).
Local physical therapies such as heat or cold can sometimes give temporary relief. Acupuncture and transcutaneous electrical nerve stimulation (TENS) have also
been shown to be effective in knee OA.
Antineuropathic drugs, such as amitriptyline, gabapentin and pregabalin, are sometimes used in patients with symptoms that are difficult to control, but the evidence base for their use is poor.
Corticosteroid injections
Intra-articular injections are effective in knee OA and are also used for symptomatic relief at the first CMC joint. The duration of effect is usually short but trials of serial corticosteroid injections every 3 months in knee OA
have shown efficacy for up to 1 year.
Chondroitin and glucosamine sulphate
Have been used alone and in combination for the treatment of knee OA. There is evidence from randomised controlledtrials that these agents can improve knee pain to a small
extent (3–5%), as compared with placebo. Although
NICE did not consider these differences to be clinically significant, the beneficial effects of paracetamol in knee OA are equally small.
Hyaluronan injections
In knee OA, intra-articular injection, given as a course of weekly injections for 3–5 weeks, may give modest pain relief for several months. However, evidence for efficacy is heterogeneous and the expense of this treatment and the common requirement for serial injection mean that injections are not recommended for OA by NICE.
Disease-modifying therapies
There are no licensed drugs that can halt the progression
of OA. Glucosamine sulphate was shown in one study
to slow the rate of radiological progression in knee OA
and to reduce the number of subjects who progressed to
joint replacement but the study has been criticised on
methodological grounds. A recent randomised controlled
trial suggested that strontium ranelate might also be
effective in reducing the rate of progression of knee OA
but it is not licensed for this indication.
Surgery
Surgery should be considered for patients with OAwhose pain, stiffness and reduced function impact significantly on their quality of life and are refractory to
other treatments. Osteotomy can prolong the life of
malaligned joints and relieve pain by reducing intraosseous pressure, but is performed infrequently. Total
joint replacement surgery is by far the most common
surgical procedure for patients with OA. It can transform the quality of life for people with severe knee or hip OA and is indicated when there is significant structural damage on X-ray, and pain and functional impairment are limiting quality of life despite the use of medical therapy.
Patient-specific factors, such as age, gender, smoking and presence of obesity, should not be barriers to referral for joint replacement. Only a small proportion of patients with OA progress to the extent that total joint replacement is required, but OA is by far the most frequent indication for a total joint replacement. Over 95% of joint replacements continue to function well into the second decade after surgery and most provide life-long, pain-free function. However, some 20% are not satisfied with the outcome, and a few experience little or no improvement in pain.
CRYSTAL-INDUCED ARTHRITIS
A variety of crystals can deposit in and around joints
and cause an acute inflammatory arthritis, as well as a more chronic arthritis associated with progressive joint .
Crystals can be the primary pathogenic agent, as in gout, or an accessory factor, as in calcium pyrophosphate deposition disease, in which crystals are deposited in joints that are already abnormal. There must be sufficient concentration of the chemical components (ionic product. The inflammatory potential of crystals resides in their physical irregularity and high negative surface charge, which can induce inflammation and damage cell membranes. Crystals may also cause mechanical damage to tissues and act as wear particles at the joint surface.
Crystals can paradoxically reside in cartilage or tendon for years without causing inflammation or symptoms, and it is only when they are released that they trigger inflammation. This may occur spontaneously but can also result from local trauma, changes in the concentration of the components that form crystals, or be part of an acute phase response due to Intercurrent illness or surgery.
Gout
Gout is an inflammatory disease caused by deposition
of monosodium urate monohydrate crystals in and
around synovial joints. The prevalence is 1–2%, with a greater than 5 : 1 male preponderance. It is the most common inflammatory arthritis in men and in older women. The risk of developing gout increases with age and with serum uric acid (SUA) levels.
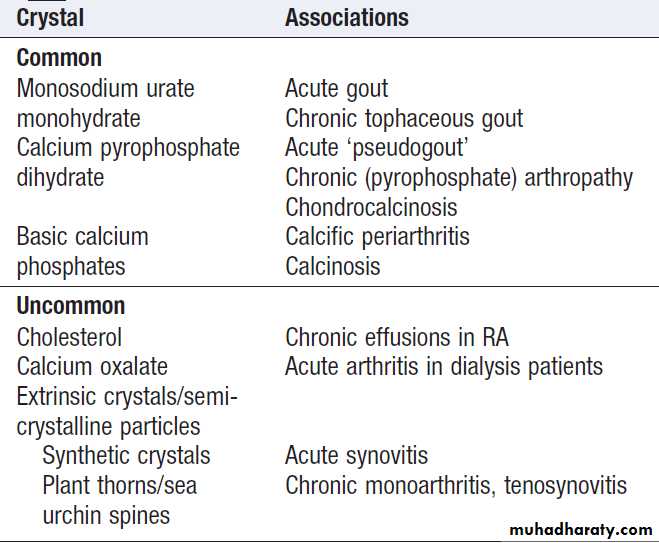
Crystal-associated arthritis and deposition
in connective tissue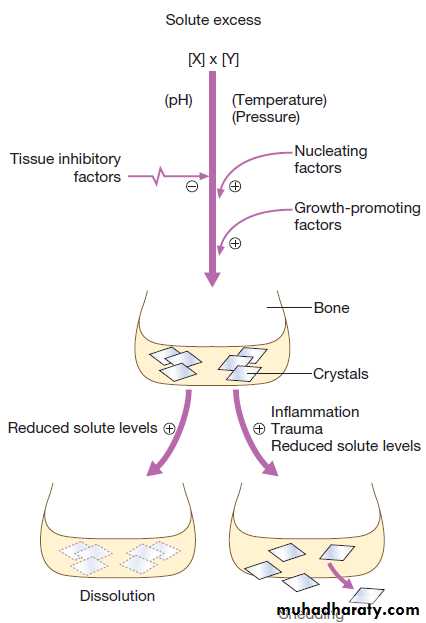
Factors affecting the balance of crystal formation and tissue concentration.
Hyperuricaemia is defined as an SUA level greater than
2 standard deviations above the mean for the population. Gout has become more common over recent years in parallel with increased longevity and the higher prevalence of metabolic syndrome, of which
hyperuricaemia is an integral component.
Although hyperuricaemia is an independent risk factor for hypertension, vascular disease, renal disease and cardiovascular events, only a minority of hyperuricaemic people develop gout.
There is currently no evidence to support the use of urate-lowering therapy in patients with asymptomatic hyperuricaemia.
Pathophysiology
About one-third of the body uric acid pool is derivedfrom dietary sources and two-thirds from endogenous
purine metabolism . The concentration of uric acid in body fluids depends on the balance between endogenous synthesis, and elimination by the kidneys (two-thirds) and gut (one-third).
Purine nucleotide synthesis and degradation are regulated by a network of enzyme pathways.
Xanthine oxidase catalyses the end conversion of hypoxanthine to xanthine and then xanthine to uric acid. In over 90% of patients, the main abnormality is reduced
uric acid excretion by the renal tubules, which impairs
the body’s ability to respond to a purine load.
In many cases, this is genetically determined and recent studies have identified polymorphisms in several genes which regulates urate excretion by the kidney.
Impaired renal excretion of urate also accounts for the
occurrence of hyperuricaemia in CKD, and for the association between hyperuricaemia and thiazide diuretics. Other risk factors include metabolic syndrome , high alcohol intake (predominantly beer, which contains guanosine), generalised OA, a diet relatively high in red meat or fructose or relatively low in vitamin C or coffee, and lead poisoning (saturnine gout).
The association between OA and gout is thought to be due to reduction in levels of proteoglycan and other inhibitors of crystal formation in osteoarthritic cartilage, predisposing to crystal formation.
Some patients develop gout because they overproduce
uric acid. The mechanisms are poorly understood,except in the case of a few rare disorders, in which
gout is inherited in a Mendelian manner as the result of
mutations in genes responsible for purine synthesis (see
Box). Lesch–Nyhan syndrome for example, is an
X-linked recessive form of gout that is also associated
with mental retardation, self-mutilation and choreoathetosis.
Severe hyperuricaemia can also occur in patients with leukaemia undergoing chemotherapy due to increased purine turnover.
Diminished renal excretion
• Increased renal tubular reabsorption* • Renal failure • Lead toxicity • Lactic acidosis • Alcohol
• Drugs Thiazide and loop diuretics . Low-dose aspirin . Ciclosporin . Pyrazinamide
Increased intake
• Red meat • Seafood • Offal
Over-production of uric acid
• Myeloproliferative and lymphoproliferative disease • Psoriasis • High fructose intake
• Glycogen storage disease • Inherited disorders Lesch–Nyhan syndrome (HPRT mutations)
Phosphoribosyl pyrophosphate synthetase 1 mutations
*Usually genetically determined (HPRT = hypoxanthine guanine phosphoribosyl transferase)
Causes of hyperuricaemia and gout
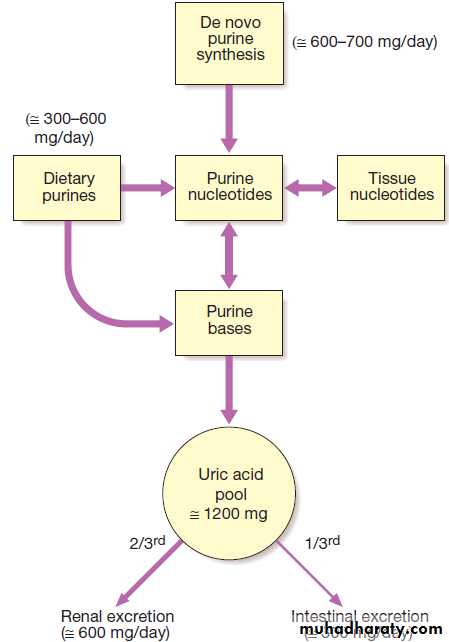
The uric acid pool. Origins and disposal of uric acid.
Clinical featuresThe classical presentation is with an acute monoarthritis,
which in over 50% of cases affects the first MTP joint.
Other common sites are the ankle, midfoot, knee, small joints of hands, wrist and elbow. The axial skeleton and large proximal joints are rarely involved.
Typical features include:
• rapid onset, reaching maximum severity in 2–6 hours, and often waking the patient in the early morning
• severe pain, often described as the ‘worst pain ever’
• extreme tenderness, such that the patient is unable
to wear a sock or to let bedding rest on the joint
• marked swelling with overlying red, shiny skin
• self-limiting over 5–14 days, with complete resolution.
During the attack, the joint shows signs of marked
synovitis, swelling and erythema. There may be accompanying fever, malaise and even confusion, especially if a large joint such as the knee is involved. As the attack subsides, pruritus and desquamation of overlying skin are common. The main differential diagnosis is septic arthritis, infective cellulitis or reactive arthritis. Acute attacks may also manifest as bursitis, tenosynovitis or cellulitis, which have the same clinical characteristics. Many patients describe milder episodes lasting just a few days. Some have attacks in more than one joint. Others have further attacks in other joints a few days later (cluster attacks), the first possibly acting as a trigger. Simultaneous polyarticular attacks are unusual.
Some people never have a second episode after an
acute attack. In others, several years may elapse beforethe next attack. In many, however, a second attack
occurs within 1 year and may progress to chronic gout,
with chronic pain and joint damage, and occasionally
severe deformity and functional impairment. Patients
with uncontrolled hyperuricaemia who suffer multiple
attacks of acute gout may also progress to chronic gout. Crystals may be deposited in the joints and soft
tissues to produce irregular firm nodules called tophi.
These have a predilection for the extensor surfaces of
fingers, hands, forearm, elbows, Achilles tendons and
sometimes the helix of the ear. Tophi have a white
colour, differentiating them from rheumatoid nodules.
Tophi can ulcerate, discharging white gritty material, become infected or induce a local inflammatory response, with erythema and pus in the absence of secondary infection. They are usually a feature of long-standing gout but can sometimes develop within 12 months in patients with CKD. Occasionally, tophi may develop in the absence of previous acute attacks, especially in patients on thiazide therapy who have coexisting OA.
Chronic hyperuricaemia may also be complicated by renal
stone and, if severe, renal impairment due to the development of interstitial nephritis as the result of urate deposition in the kidney.
This is particularly common in patients with chronic tophaceous gout who are on diuretic therapy.
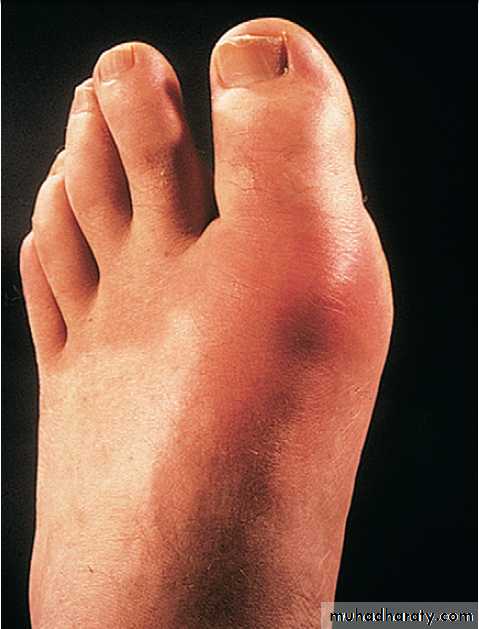
Podagra. Acute gout causing swelling, erythema and extreme pain and tenderness of the first metatarsophalangeal jt.
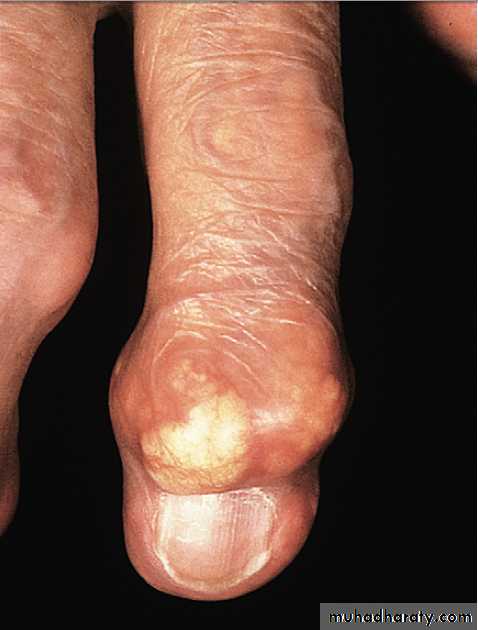
Tophus with white monosodium urate monohydrate crystals visible beneath the skin. Diuretic-induced gout in a patient with pre-existing nodal OA.
Investigations
The diagnosis of gout can be confirmed by the identification of urate crystals in the aspirate from a joint, bursa or tophus . In acute gout, synovial fluid shows increased turbidity due to the greatly elevated cell count (> 90% neutrophils). In chronic gout, the appearance is more variable but occasionally the fluid appears white due to the high crystal load. Between attacks, aspiration of an asymptomatic first MTP joint or knee may still reveal crystals. A biochemical screen, including renal function, uric acid, glucose and lipid profile, should be performed because of the association with metabolic syndrome. Although hyperuricaemia is usually present, this does not confirm the diagnosis.Conversely, normal uric acid levels during an attack do not exclude gout, as serum urate falls during the acute phase response. Elevated ESR and CRP and a neutrophilia are typical of acute gout, and they return to normal as the attack subsides. Tophaceous gout may be accompanied by a modest but chronic elevation in ESR and CRP. Radiographs are usually normal in acute gout but
well-demarcated erosions may be seen in patients with
chronic or tophaceous gout .
Tophi may also be visible on X-rays as soft tissue swellings.
In late disease, destructive changes may occur similar to those in other forms of advanced inflammatory arthritis.
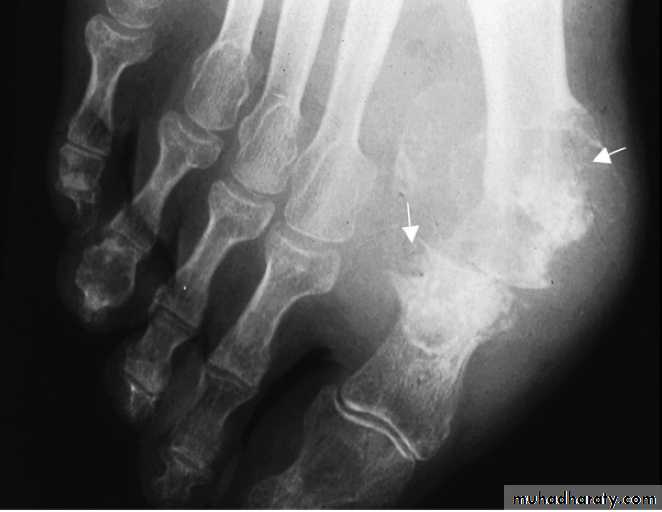
Erosive arthritis in chronic gout. Punched-out erosions
are visible (arrows), in association with a destructive arthritis affecting the first metatarsophalangeal joint.Management
Oral NSAIDs are effective for pain relief in the acute
attack and are the standard treatment, but have to be prescribed with caution in old age. Local ice packs can
also be used for symptomatic relief. Patients with recurrent
episodes can keep a supply of an NSAID and take
it as soon as the first symptoms occur, continuing until
the attack resolves.
Oral colchicine, which works by inhibiting microtubule assembly in neutrophils, is also very effective. It is usually given in doses of 0.5 mg twice or 3 times daily. The most common adverse effects are nausea, vomiting and diarrhoea. Joint aspiration can give pain relief, and may be combined with an intraarticular steroid injection if the diagnosis is clear and infection can be excluded.
A short course of oral or intramuscular corticosteroids can also be highly effective in treating acute attacks.
Patients who have more than one acute attack within
12 months and those with complications should be
offered urate-lowering therapy .The longterm
therapeutic aim is to prevent attacks occurring by
bringing uric acid levels below the level at which monosodium urate monohydrate crystals form.
A therapeutic target of 360 μmol/L (6 mg/dL) is recommended. Allopurinol is the drug of first choice. It is a xanthine
oxidase inhibitor, which reduces the conversion of
hypoxanthine and xanthine to uric acid. The recommended
starting dose is 100 mg daily, or 50 mg in older
patients and in renal impairment.
The dose of allopurinol should be increased by 100 mg every 4 weeks (50 mg in the elderly and those with renal impairment) until the target uric acid level is achieved, side-effects occur or the maximum recommended dose is reached (900 mg/day).
Acute flares of gout often occur following initiation of
urate-lowering therapy. The patient should be warned
about this and told to continue therapy, even if an attack
occurs. The risk of flares can be reduced by administration
of oral colchicine (0.5 mg twice daily) or NSAID
therapy for the first few months. In the longer term,
annual monitoring of uric acid levels is recommended.
In most patients, urate-lowering therapy needs to be
continued indefinitely.
Febuxostat is a xanthine oxidase inhibitor that is
useful in patients who fail to respond adequately toallopurinol, and those in whom it is contraindicated or
has been poorly tolerated. It undergoes hepatic metabolism and so no dose adjustment is required for renal impairment. It is more effective than allopurinol at reducing uric acid levels and, as a result, commonly provokes attacks at the recommended starting dose (80 mg daily). In view of this, treatment with colchicine or NSAID should be considered for the initial 6 months.
Uricosuric drugs, such as probenecid or sulfinpyrazone,
can be effective but require several doses each
day and maintenance of a high urine flow to avoid uric acid crystallisation in renal tubules.
Salicylates antagonise the uricosuric action of these drugs and should be avoided.
Uricosurics are contraindicated in over-producers, those with renal impairment and in urolithiasis (they increase stone formation). The uricosuric benzbromarone can be very effective and safe in mild to moderate renal impairment, but can cause hepatotoxicity.It is not licensed in the UK for the treatment of
hyperuricaemia.
Pegloticase is a biological treatment in which the enzyme uricase has been conjugated to monomethoxypolyethylene
glycol. It is indicated for the treatment of tophaceous gout resistant to standard therapy and is administered as an intravenous infusion every 2 weeks for up to 6 months.
It is highly effective at controlling hyperuricaemia and causes regression of tophi. The main adverse effects are infusion reactions (which can be treated with antihistamines or steroids) and flares of gout during the first 3 months of therapy.
A limiting factor for longer-term treatment is the development of antibodies to pegloticase, which occur in a high proportion of cases and are associated with an impaired therapeutic response. In addition to drug treatment, predisposing factors
should be corrected if possible. Patients should be
advised to lose weight where appropriate and to reduce
excessive alcohol intake, especially beer.
Thiazide diuretics should be stopped if possible and substituted with angiotensin-converting enzyme (ACE) inhibitors, as these have a uricosuric effect.
Patients should be advised to avoid large amounts of seafood and offal, which have a high purine content, but a highly restrictive diet is not necessary.

Indications for urate-lowering drugs

Gout in old age
Calcium pyrophosphate dehydrate crystal deposition disease ( pseudogout)This condition is associated with deposition of calcium
pyrophosphate dihydrate (CPPD) crystals within articular and hyaline cartilage and is sometimes known as ‘pseudogout’. It is rare under the age of 55. The knee (hyaline cartilage and menisci) is by far the most common site, followed by the wrist (triangular fibrocartilage) and pelvis (symphysis pubis). In many patients, chondrocalcinosis is asymptomatic and an incidental finding on X-ray, but others present with an acute inflammatory arthritis (pseudogout) or a chronic inflammatory arthropathy superimposed on a background of OA, especially at the knee associated with joint damage and limitation.

Risk factors for chondrocalcinosis
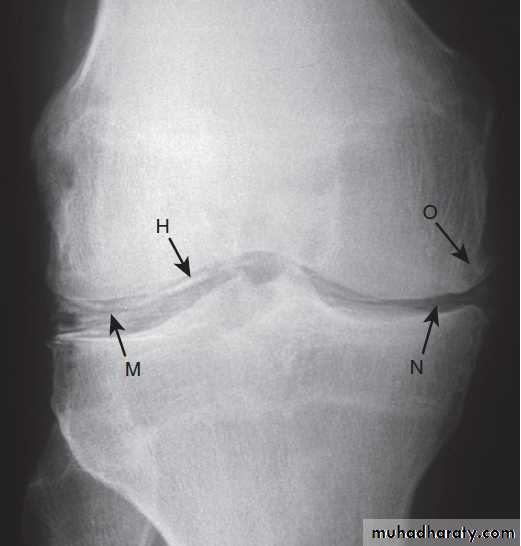
Chondrocalcinosis of the knee. The X-ray shows
calcification of the fibrocartilaginous menisci (M) and articular hyalinecartilage (H). There is also narrowing (N) and osteophyte (O) of the medial
tibio-femoral compartment.
Pathophysiology
The underlying mechanisms of crystal deposition arepoorly understood. Clinical studies have shown that
pyrophosphate levels are raised in patients with
CPPD crystal deposition disease, possibly due to
over-production, but why this occurs is unclear. In
hypophosphatasia, the predisposing factor is thought to
be impaired degradation of pyrophosphate due to mutations in the genes that encode ALP.
OA is thought to predispose to CPPD crystal deposition disease because of a reduction in the amounts of proteoglycan and other natural inhibitors of crystal formation in the abnormal cartilage.
Clinical features
A common presentation is with an acute inflammatory
arthritis that resembles acute gout, sometimes termed
‘pseudogout’. Examination reveals a warm, tender erythematous joint with signs of a large effusion. Fever
is common and the patient may appear confused and ill.
The knee is most commonly affected, followed by the
wrist, shoulder, ankle and elbow.
Trigger factors include trauma, intercurrent illness and surgery.
Sepsis and gout are the main differential diagnoses. Chronic arthropathy may also occur in association with CPPD crystal deposition disease, affecting the same joints that are involved in acute pseudogout.
The presentation is with chronic pain, early morning stiffness and functional impairment. Acute
attacks of pseudogout may be superimposed. Affected
joints usually show features of OA, with varying degrees
of synovitis. Effusion and synovial thickening are usually most apparent at knees and wrists; wrist involvement may result in carpal tunnel syndrome. Inflammatory features may be sufficiently pronounced to suggest RA, but tenosynovitis and extra-articular involvement are absent, and large and medium rather than small
joints are targeted.
Severe damage and instability of knees or shoulders may occasionally lead to consideration of a neuropathic joint, but no neurological abnormalities will be found.
Investigations
Examination of synovial fluid using compensated polarized microscopy will demonstrate CPPD crystals and permit distinction from gout. The aspirated fluid is often turbid and may be uniformly blood-stained, reflecting the severity of inflammation. Since sepsis and pseudogout can coexist, Gram stain and culture of the fluid should be performed to exclude sepsis, even if CPPD crystals are identified in synovial fluid. X-rays of the affected joint may show evidence of calcification in hyaline cartilage and/or fibrocartilage, although absence of calcification does not exclude the diagnosis. Signs of OA are frequently present. Screening for secondary causes should be undertaken, especially in patients who present under the age of 25 and those with polyarticular disease.Management
Joint aspiration can often provide symptomatic relief inpseudogout and sometimes no further treatment is
required. Patients with persistent symptoms can be
treated with intra-articular corticosteroids, colchicine
or NSAID. Since most patients with pseudogout are
elderly, NSAID must be used with caution. Early active
mobilisation is also important.
Chronic pyrophosphate-induced arthropathy should be managed as for OA .
Calcific periarthritis
Deposition of BCP in tendons may result in an acute
inflammatory response. The most commonly affected
site is the supraspinatus tendon but other sites may also be affected, including the tendons around the hip, feet and hands. The presentation is with acute pain, swelling and local tenderness that develops rapidly over 4–6 hours. The overlying skin may be hot and red, raising the possibility of infection. Attacks sometimes occur spontaneously but can also be triggered by trauma. Modest systemic upset and fever are common. X-rays show tendon calcification. If the affected joint or bursa is aspirated, inflammatory fluid containing many calcium-staining (alizarin red S) aggregates may be obtained.
During an acute attack, there may be a neutrophilia
with an elevation in ESR and CRP. Routinebiochemistry is normal.
Treatment is with analgesics and NSAID. Attacks may also respond to a local injection of corticosteroid. The condition usually resolves spontaneously over 1–3 weeks and this is often accompanied by dispersal and disappearance of small to medium-sized BCP deposits on X-ray. Large deposits sometimes accumulate, causing limitation of joint movement, and may require surgical removal.
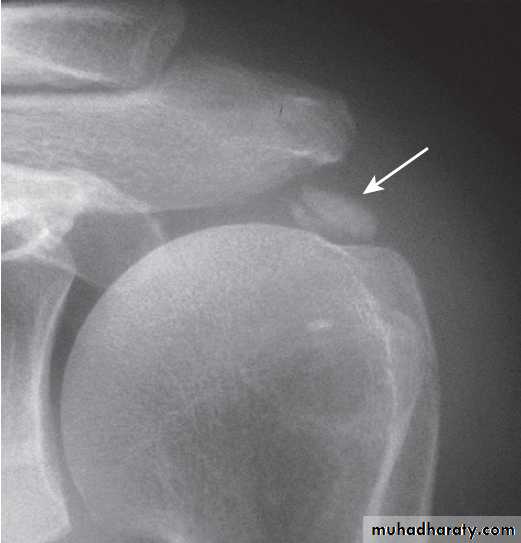
Shoulder X-ray showing supraspinatus tendon
calcification (arrow).FIBROMYALGIA
This is a common cause of generalised regional pain anddisability, and is frequently associated with medically
unexplained symptoms in other systems .The
prevalence in the UK and US is about 2–3%. Although
fibromyalgia can occur at any age, including adolescence,
it increases in prevalence with age, to reach a peak
of 7% in women aged over 70. There is a strong female predominance of around 10 : 1. Risk factors include life
events that cause psychosocial distress, such as marital
disharmony, alcoholism in the family, injury or assault,
low income and self-reported childhood abuse.

Predisposing factors for fibromyalgia
PathophysiologyThe cause of fibromyalgia is poorly understood but two
abnormalities that may be interrelated have
been consistently reported in affected patients.
Sleep abnormality
Delta waves are characteristic of deep stages of
non-rapid eye movement (non-REM) sleep, usually
occurring in the first few hours and thought to have an
important restorative function. People with fibromyalgia
have reduced delta sleep in a pattern distinct from
that seen with depression. Furthermore, deprivation of
delta but not REM sleep in normal volunteers produces
the symptoms and signs of fibromyalgia, supporting it
as a non-restorative sleep disorder.
Abnormal peripheral and central pain processing
A reduced threshold of pain perception and tolerance atcharacteristic sites throughout the body is characteristic .
Affected people have peripheral sensitization and spinal cord ‘wind-up’ (pain amplification),with an exaggerated skin flare response to topically applied capsaicin, and frequent occurrence of dermatographism and allodynia (normally non-noxious stimuli become painful). Abnormal central pain processing is suggested by altered CSF levels of substance P (increased) and serotonin (reduced); reduced basal levels of regional cerebral blood flow in the caudate and thalamus; low basal free cortisol.
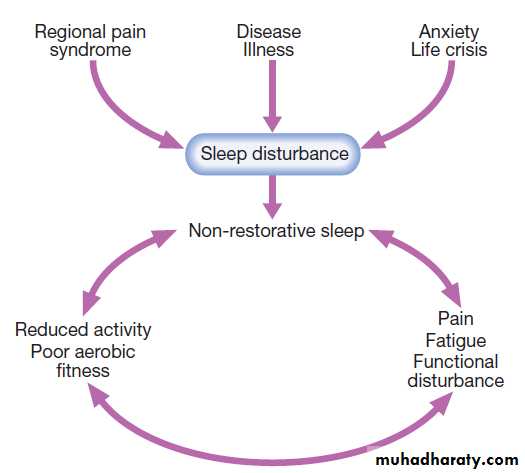
Possible causative mechanisms in fibromyalgia.
Clinical featuresThe main presenting feature is widespread pain, which
is often worst in the neck and back .The pain
is characteristically diffuse and unresponsive to analgesics and NSAID. Physiotherapy often makes it worse.
Fatiguability, most prominent in the morning, is another
major problem and disability is often marked. Although
people can usually dress, feed and groom themselves, they may be unable to perform tasks such as shopping
or housework.
They may have experienced major difficulties at work or even retire because of pain and fatigue.
Examination is unremarkable, apart from the presence
of hyperalgesia on moderate digital pressure inmultiple sites . The tenderness can be quantitated
using metered dolorimeters but moderate digital
pressure, enough just to whiten the nail, is sufficient for
clinical diagnosis. Patients with other musculoskeletal
diseases can develop fibromyalgia and it is important to
determine to what extent symptoms are related to fibromyalgia or a coexisting disease.
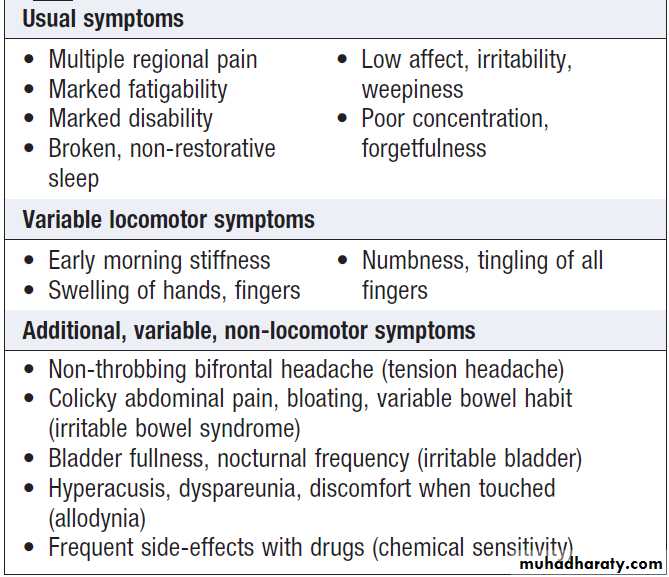
Clinical features in fibromyalgia
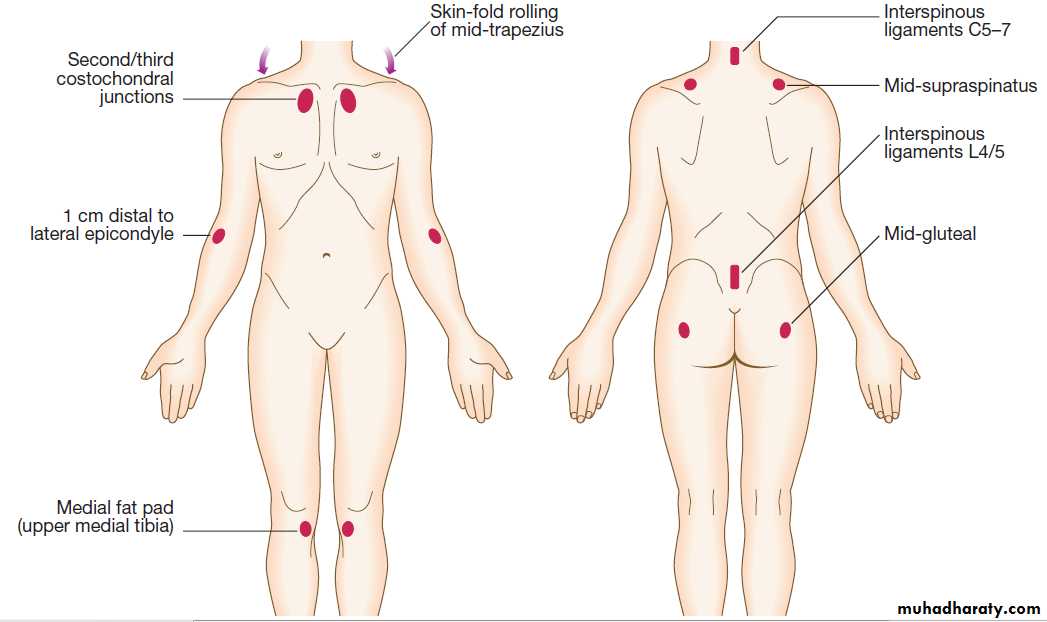
Typical tender spots in fibromyalgia.
Investigations and managementThere are no abnormalities on routine blood tests or
imaging, but it is important to screen for other conditions
that could contribute to some of the patient’s symptoms .
The aims of management are to educate the patient
about the condition, achieve pain control and improve
sleep. Wherever possible, education should include the spouse, family or carer. The model of a self-perpetuating cycle of poor sleep and pain is a useful framework for problem-based management. Understanding the diagnosis can often help the patient come to terms with the symptoms.
Repeat or drawn-out investigation may reinforce beliefs in occult serious pathology and should be avoided.
Low-dose amitriptyline (10–75 mg at night), with or
without fluoxetine, may help by encouraging delta sleepand reducing spinal cord wind-up. Many people with
fibromyalgia, however, are intolerant of even small
doses of amitriptyline. There is limited evidence for
the use of tramadol, serotonin–noradrenaline (norepinephrine) re-uptake inhibitors (SNRIs) such as duloxetine, and the anticonvulsants pregabalin and gabapentin.
A graded increase in aerobic exercise can improve wellbeing and sleep quality. The use of self-help strategies
and a cognitive behavioural approach with relaxation
techniques should be encouraged.
Sublimated anxiety relating to distressing life events should be specifically explored with appropriate counselling. There are patient organisations that provide additional information and support.
The prognosis for hospital-diagnosed fibromyalgia is
poor. Although treatment may improve quality of life
and ability to cope, most people do not lose their symptoms over 5 years. Subjects diagnosed in primary care, or who have sublimated anxiety that can be successfully addressed, may fare better.
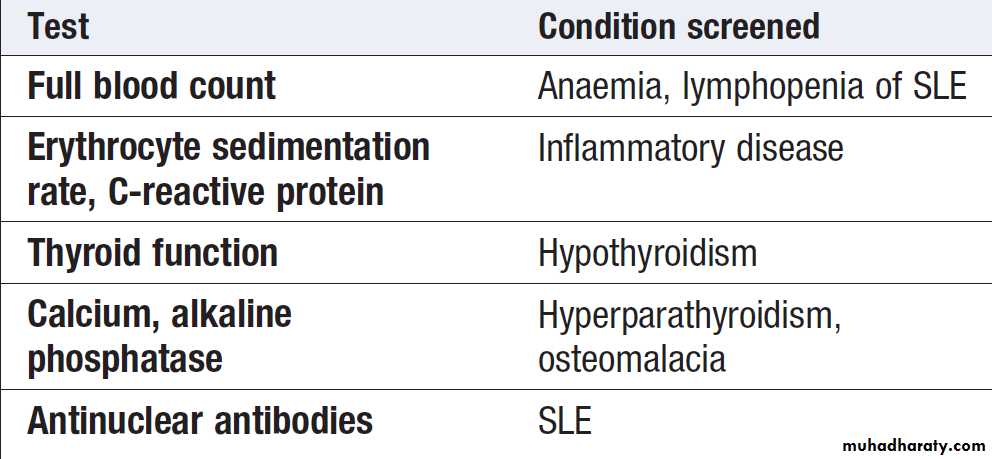
Minimum investigation screen in fibromyalgia
BONE AND JOINT INFECTIONSeptic arthritis
The most rapid and destructive joint disease, associated with significant morbidity and a mortality of 10%. This has not improved over the last 20 years, despite advances in antimicrobial therapy. The incidence is 2–10 per 100 000 in the general population, and 30–70 per 100 000 in those with pre-existing joint disease or joint replacement. Usually due to haematogenous spread from either skin or upper respiratory tract; infection from direct puncture wounds or secondary to joint aspiration is uncommon. Risk factors include increasing age, pre-existing joint disease (principally RA), diabetes, immunosuppression and IV drug misuse. In RA, the skin is a frequent portal of entry because of maceration of skin between the toes due to joint deformity
Clinical features
The usual presentation is with acute or subacute monoarthritis and fever. The joint is usually swollen, hotand red, with pain at rest and on movement. Although any joint can be affected, lower limb joints, knee and hip, are commonly targeted. Patients with pre-existing arthritis may present with multiple joint involvement. In adults, the most likely organism is Staphylococcus aureus, particularly in patients with RA and diabetes. In sexually active adults, disseminated gonococcal infection occurs in up to 3% of untreated gonorrhoea, usually presenting with migratory arthralgia, low-grade fever and tenosynovitis, which may precede the development of oligo- or monoarthritis. Painful pustular skin lesions may also be present.
Amongst the elderly and IV drug users, Gram-negative bacilli or group B, C and G streptococci are important causes. Group A streptococci, pneumococci, meningococci and Haemophilus influenzae are occasionally isolated.
Investigations
The pivotal investigation is joint aspiration but blood
cultures should also be taken. The synovial fluid is
usually turbid or blood-stained but may appear normal.
If the joint is not readily accessible, aspiration should be
performed under imaging guidance or in theatre. Prosthetic joints should only be aspirated in theatre. Synovial fluid should be sent for Gram stain and
culture; cultures are positive in around 90% of cases, but
the Gram stain is positive in only 50%.
In contrast, synovial fluid culture is positive in only 30% of gonococcal infections, making it important to obtain concurrent cultures from the genital tract (positive in 70–90% of cases). There is a leucocytosis with raised ESR and CRP in most patients, but these features may be absent in elderly or immunocompromised patients, or early in the disease
course. Serial measurements of CRP and ESR are useful
in following the response to treatment.
Management
The patient should be admitted to hospital for pain relief and administration of parenteral antibiotics. Flucloxacillin (2 g IV 4 times daily) is the antibiotic of first choice pending the results of cultures, since it will cover most staphylococcal and streptococcal infections.
If there is reason to suspect a Gram-negative infection, then a cephalosporin or gentamicin should be added. Microbiology advice should be sought in complicated situations such as intravenous drug users, patients in intensive care and those who might be colonised by resistant organisms. It is traditional to continue intravenous antibiotics for 2 weeks and to follow this with oral treatment for another 4 weeks, but there is no evidence to support the optimal duration of treatment. Joint aspiration should be performed using a large-bore needle once or twice daily. If this is not possible, arthroscopic or open surgical drainage may need to be undertaken.
Regular passive movement should be undertaken from the outset, and active movements encouraged once the condition has stabilised. Infected prosthetic joints require management by the orthopaedic team, but often prolonged antibiotic treatment on its own is ineffective and removal of the prosthesis is required for eradication of the infection.
Brucellosis presents with an acute febrile illness, followed in some cases by the development of localised infection, which can result in arthritis, bursitis, osteomyelitis, sacroiliitis and paravertebral or psoas abscesses.
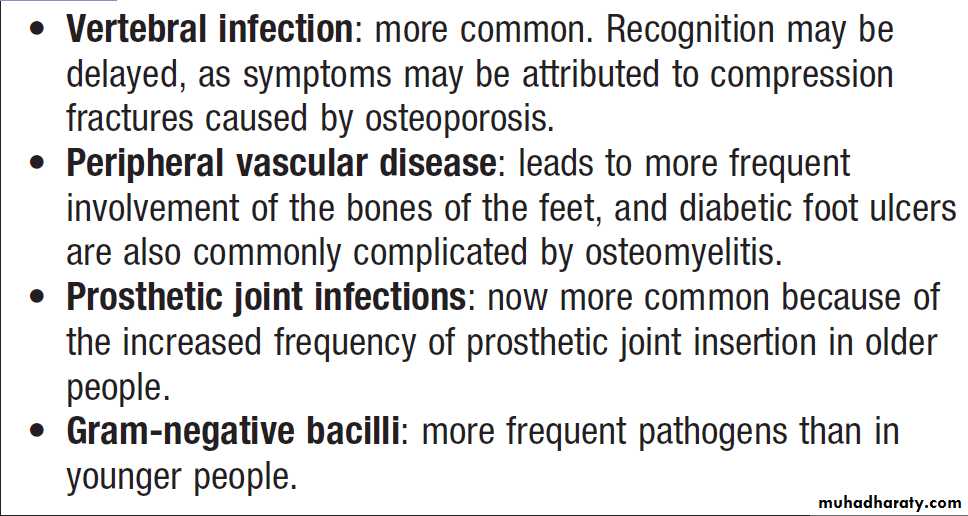
Joint and bone infection in old age
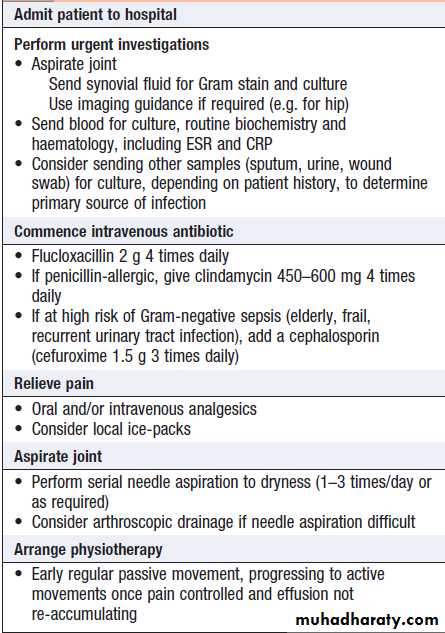
Emergency management of suspected
septic arthritisViral arthritis
The usual presentation is with acute polyarthritis, following
a febrile illness, which may be accompanied by
a rash. Most cases of viral arthritis are self-limiting and
settle down within 4–6 weeks. Human parvovirus
arthropathy is the most common in Europe; adults may not have the characteristic ‘slapped cheek’ facial rash seen in children. The diagnosis can be confirmed by a rise in specific IgM. Polyarthritis may also occur rarely with hepatitis B and C, rubella (including rubella vaccination) and HIV infection. A variety of mosquito-borne viruses may cause epidemics of acute polyarthritis, and HIV,
Management
is symptomatic, with NSAID and analgesics

Musculoskeletal manifestations of HIV
OsteomyelitisIn osteomyelitis, the primary site of infection is bone and
bone marrow. Any part of a bone may be involved but
there is preferential targeting of the juxta-epiphyseal
regions of long bones adjacent to joints. The usual source
is through haematogenous spread, although directly
introduced infection may complicate trauma or orthopaedic surgery. The organisms most frequently implicated are staphylococci, Pseudomonas and Mycobacterium tuberculosis. Osteomyelitis occurs most commonly in childhood and adolescence. Risk factors include diabetes mellitus ,compromised immunity (including AIDS) and sickle cell disease, which particularly increases the risk of Salmonella infection.
The infection often results in a florid inflammatory
response, with a greatly increased intraosseous pressure.
If untreated, the condition may cause localised
areas of osteonecrosis, leading to the development of a
fragment of necrotic bone that is called a sequestrum.
Eventual perforation of the cortex by pus stimulates
local new bone formation (involucrum) in the periosteum,
often leading to the development of sinuses that
discharge through the skin.
Clinical features and investigations
The presentation is with localised bone pain and tenderness, often accompanied by malaise, night sweats and pyrexia.The adjacent joint may be painful to move and
may develop a sterile effusion or secondary septic arthritis.
X-rays may show evidence of osteopenia, localised
osteolysis and osteonecrosis, but the imaging modality
of choice is MRI, which is much more sensitive for
detecting early changes. Cultures should be performed,
where possible, by open or imaging-guided biopsy.
Blood cultures should be taken, which may also reveal
the causative organism.
Management
Early recognition and management is critical; once
osteomyelitis becomes established and chronic, it
may prove very hard to eradicate the infection with antibiotics alone. The principles are those followed for
septic arthritis, with parenteral antibiotics for at least
2 weeks, followed by oral antibiotics for at least 4 weeks.
Resection of the infected bone and subsequent reconstruction are often required.
Complications of chronic osteomyelitis include secondary amyloidosis and skin malignancy at the margin of a discharging sinus (Marjolin’s ulcer).
Tuberculosis
Tuberculosis can affect the musculoskeletal system,usually targeting the spine (Pott’s disease) or large joints
such as the hip, knee or ankle. The presentation is with
pain, swelling and fever. The X-ray changes are nonspecific and mycobacteria are seldom identified in the
synovial fluid, so tissue biopsy is required for a definite
diagnosis. In some cases, surgical débridement may be required for extensive joint disease, and spinal involvement may require surgical stabilisation and decompression.
RHEUMATOID ARTHRITIS (RA)
Is the most common persistent inflammatory arthritis, occurring throughout the world and in all ethnic groups. The prevalence is lowest in black Africans and Chinese, and highest in Pima Indians. In Caucasians, approximately 0.8–1.0% are affected, with a female to male ratio of 3 : 1. The clinical course is prolonged, with intermittent exacerbations and remissions. Patients with RA have an increased mortality when compared with age-matched controls, primarily due to an increased risk of cardiovascular disease. This is most marked in those with severe disease, with a reduction in expected lifespan by 8–15 years. Around 40% of RA patients are registered as disabled within 3 years of onset, and around 80% are moderately to severely disabled within 20 years.Functional capacity
decreases most rapidly at the beginning of disease andthe functional status of patients within their first year of
RA is often predictive of long-term outcome. Factors that
associate with a poorer prognosis are disability at presentation, female gender, involvement of MTP joints,
radiographic damage at presentation, smoking and a
positive RF or ACPA. It is likely that the prognosis of
RA will improve as more effective treatment regimens
are introduced in patients with early disease. In former
years, around 25% of patients required a large joint
replacement but rates are now falling, probably reflecting
more aggressive and effective medical therapy.
Pathophysiology
Genetic, epigenetic and environmental factors are implicated in the pathogenesis of RA. The concordance
rate of RA is higher in monozygotic (12–15%) than in
dizygotic twins (3%), and there is an increased frequency
of disease in first-degree relatives of patients. Genomewide association studies have detected more than 30 loci that are associated with the risk of developing RA or with severity of disease once it is established. Many of the risk genes are involved in the function of the immune
system, and include major histocompatibility complex
(MHC) class II, PTPN22, CD40L and CTLA4.
The MHC class II gene, HLA-DR4, is the major susceptibility haplotype in most ethnic groups, occurring in 50–75% of Caucasian patients with RA, compared to 20–25% of the
normal population. However, DR1 is more important in
Indians and Israelis, and DW15 in Japanese. It has long
been thought that RA may be triggered by an infectious
agent in a genetically susceptible host, but a specific
pathogen has not been identified. Periodontal disease
and oral pathogens have been implicated, as have GI organisms, and viruses such as Epstein– Barr and CMV. Cigarette smoking is a strong risk factor for developing RA, especially in people with HLA-DR4, and is also associated with greater disease severity and reduced responsiveness to DMARD and biological treatment.
Susceptibility is increased postpartum and by breastfeeding, indicating that hormone/ immune interactions may be important.
The clinical onset of RA is characterised by infiltration
of the synovial membrane with lymphocytes, plasma
cells, dendritic cells and macrophages. CD4+ T lymphocytes, including Th1 cells (interferon-gamma (IFN-γ)
producers) and Th17 cells (IL-17A, IL-17F and IL-22 producers), play a central role by interacting with other cells in the synovium. Lymphoid follicles form within the
synovial membrane in which T cell–B cell interactions
lead B cells to produce cytokines and autoantibodies,
including RF and ACPA.
Synovial macrophages are activated by immune complexes and local damage-associated molecules acting via Toll-like receptors, to produce proinflammatory
cytokines, including TNF, IL-1, IL-6 and IL-15. These act on synovial fibroblasts, to promote swelling of the synovial membrane and damage to soft tissues and cartilage. Fibroblasts, in particular, form a rich source of inflammatory cytokines, chemokines, leukotrienes
and matrix metalloproteinases that drive
local tissue damage and remodelling. Similarly, activation
of osteoclasts by RANKL and chondrocytes by
cytokines such as IL-1 and TNF drives destruction of
bone and cartilage respectively .
The RA joint is hypoxic and this promotes new blood vessel formation (neoangiogenesis). Thus the inflamed synovium becomes vascularised, with highly activated endothelial cells supporting the recruitment of yet more leucocytes to perpetuate the inflammatory process. Amongst the range of inflammatory mediators present in the RA joint, TNF
plays an important role by regulating production of
other cytokines, whose actions are shown in Figure,
and by activating the endothelium. IL-6 similarly plays
a critical inflammatory role within the joint and also in
regulating the systemic effects of RA by inducing the
acute phase response, anaemia of chronic disease, fatigue
and reduced cognitive function.
The inflammatory granulation tissue (pannus) formed
by the above sequence of events spreads over and underthe articular cartilage, which is progressively eroded
and destroyed. Maturation of osteoclasts in the synovial
membrane and in adjacent bone combine to erode bony
structures. Later, fibrous or bony ankylosis may occur.
Muscles adjacent to inflamed joints atrophy and may be
infiltrated with lymphocytes.
This leads to progressive biomechanical dysfunction and may further amplify destruction.
Rheumatoid nodules consist of a central area of fibrinoid
material surrounded by a palisade of proliferating
mononuclear cells.
Similar granulomatous lesions may occur in the pleura, lung, pericardium and sclera. Lymph nodes in RA are often hyperplastic, showing many lymphoid follicles with large germinal centres and numerous plasma cells in the sinuses and medullary cords.
Immunofluorescence confirms RF and ACPA synthesis
by plasma cells in synovium and lymph nodes. The bone
marrow in RA is similarly hyperplastic, with evidence
of lymphocyte activation and proliferation and local
cytokine production. The bone marrow forms a selective
survival niche for plasma cells that, in turn, produce
autoantibodies. Importantly, these plasma cells are
CD20-negative and, as such, escape therapies that target
CD20, such as rituximab .
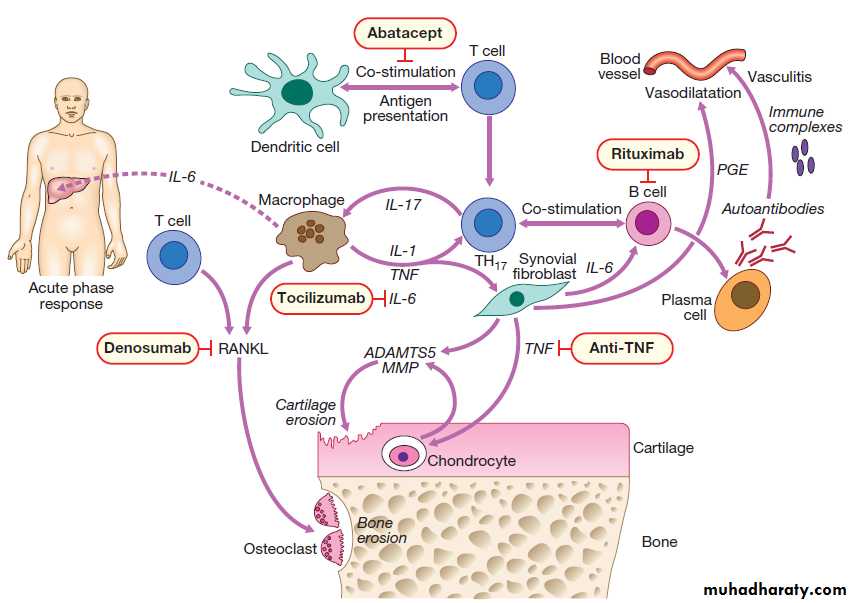
Pathophysiology of rheumatoid arthritis. Some of the cytokines and cellular interactions believed to be important in RA are shown, along
with the targets for currently available biological drugs. (ADAMTS5 = aggrecanase; IL = interleukin; MMP = matrix metalloproteinases; PGE = prostaglandin E; TNF = tumour necrosis factor; RANKL = RANK ligand)
Clinical features
The typical presentation is with pain, joint swelling andstiffness affecting the small joints of the hands, feet and
wrists.
Large joint involvement, systemic symptoms and extra-articular features may also occur. Clinical criteria for the diagnosis of RA are shown in Box. Sometimes RA has a very acute onset, with florid morning stiffness, polyarthritis and pitting oedema. This occurs more commonly in old age. Other patients may present with proximal muscle stiffness mimicking polymyalgia rheumatica . Occasionally, the onset is palindromic, with relapsing and remitting episodes of pain, stiffness and swelling that last for only a
few hours or days.
Examination reveals typical features of symmetrical
swelling of the MCP and PIP joints. These and other joints
are tender on pressure when actively inflamed and have
stress pain on passive movement. Erythema is unusual
and its presence suggests coexistent sepsis. Characteristic
deformities may develop with long-standing uncontrolled
disease, including ‘swan neck’, boutonnière , ‘button hole’ deformity, and a Z deformity of the thumb . Dorsal subluxation of the ulna at the distal radio-ulnar joint is common and may contribute to rupture of the fourth and fifth extensor tendons.
Triggering of fingers may occur because of nodules in the
flexor tendon sheaths.
Deformities of the hand are less common now, as a result of more aggressive treatment.
In the foot, dorsal subluxation of the MTP joints mayresult in ‘cock-up’ toe deformities. This causes pain on
weight-bearing on the exposed MTP heads and development of secondary adventitious bursae and callosities.
In the hindfoot, calcaneovalgus (eversion) is the most
common deformity, reflecting damage to the ankle and
subtalar joint. This is often associated with loss of the
longitudinal arch (flat foot) due to rupture of the tibialis
posterior tendon.
Popliteal (‘Baker’s’) cysts may occur in combination
with knee synovitis, where synovial fluid communicateswith the cyst but is prevented from returning to the joint
by a valve-like mechanism. Rupture, often induced by knee flexion in the presence of a large effusion, leads to calf pain and swelling that may DVT.
Extra-articular features
Anorexia, weight loss and fatigue are common and
may occur throughout the disease course. Generalised
osteoporosis and muscle-wasting result from systemic
inflammation. Extra-articular features are most common
in patients with long-standing seropositive erosive
disease but may occasionally occur at presentation,
especially in men. Most are due to serositis, granuloma
and nodule formation or vasculitis .
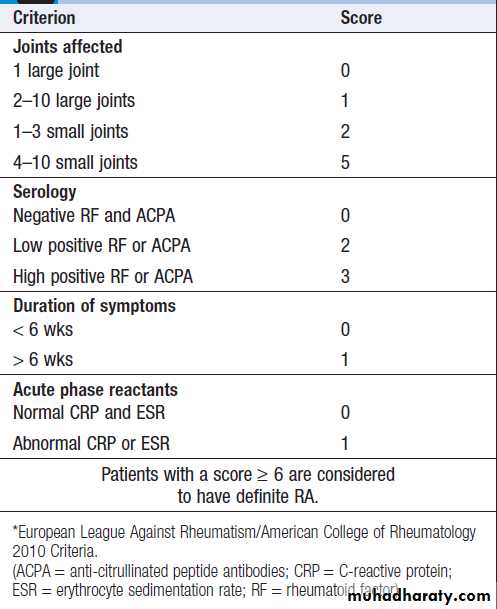
Criteria for diagnosis of
rheumatoid arthritis*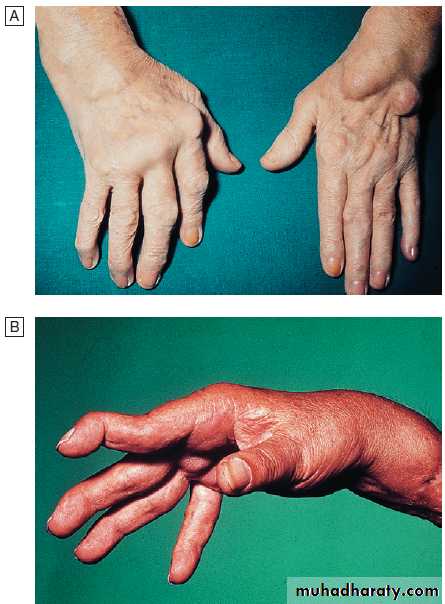
The hand in rheumatoid arthritis.
A Ulnar deviationof the fingers with wasting of the small muscles of the hands and
synovial swelling at the wrists, the extensor tendon sheaths, the
metacarpophalangeal and proximal interphalangeal joints.
B ‘Swan neck’
deformity of the fingers.
Systemic
• Fever • Weight loss • Fatigue • Susceptibility to infectionMusculoskeletal
• Muscle-wasting • Tenosynovitis • Bursitis • Osteoporosis
Haematological
• Anaemia • Thrombocytosis • Eosinophilia
Lymphatic
• Felty’s syndrome • Splenomegaly
Nodules
• Sinuses • Fistulae
Ocular
• Episcleritis • Scleritis • Scleromalacia • Keratoconjunctivitis sicca
Vasculitis
• Digital arteritis • Ulcers • Pyoderma gangrenosum • Mononeuritis multiplex•Visceral arteritis
Cardiac
• Pericarditis • Myocarditis • Endocarditis • Conduction defects • Coronary vasculitis • aortitis
Pulmonary
• Nodules • Pleural effusions • Fibrosing alveolitis • Bronchiolitis • Caplan’s syndrome
Neurological
• Cervical cord compression • Compression neuropathies • Peripheral neuropathy
• Mononeuritis multiplex
Amyloidosis
Extra-articular manifestations of rheumatoid disease
Cutaneous and vascular features
Rheumatoid nodules occur almost exclusively in seropositive patients, usually at sites of pressure or friction, such as the extensor surfaces of the forearm ,
sacrum, Achilles tendon and toes. They may be complicated by ulceration and secondary infection. Rheumatoid vasculitis may occur in older seropositive patients and is indicated by systemic symptoms (fatigue, fever) and multiple extra-articular features. Vasculitis can vary from relatively benign nail-fold infarcts to widespread cutaneous ulceration and skin necrosis. Rarely, involvement of medium-sized arteries can lead to mesenteric, renal or coronary artery occlusion.
Rheumatoid nodules and olecranon bursitis. Nodules
were palpable within as well as outside the bursa.Ocular involvement
The most common symptom is dry eyes (keratoconjunctivitissicca) due to secondary Sjِgren’s syndrome.
Painless episcleritis frequently accompanies nodular seropositive disease; it may cause intense redness of superficial vessels but sight is unimpaired.
Scleritis is more serious and potentially sight-threatening;
the eye is red and painful, vision is impaired, and
the sclera shows a deep red blush beneath the
individual red superficial vessels . Scleromalacia
is painless bilateral thinning of the sclera, with the
affected area appearing blue or grey (the colour of the
underlying choroid).
Corneal melting is a rare but devastating manifestation. It occurs in long-standing disease and is associated with systemic vasculitis. It causes pain, redness and blurred vision with corneal thinning. If it is untreated, progression to perforation is common, so urgent high-dose steroid and immunosuppressive therapy is indicated.
Cardiac and pulmonary involvement
Cardiac involvement occurs in up to 30% with seropositive RA but is usually asymptomatic. Symptomatic pericardial effusions and constrictive pericarditis are rare. Occasionally, granulomatous lesions can cause heart block, cardiomyopathy, coronary artery occlusion or aortic regurgitation. Serositis is commonly asymptomatic but may present as pleurisy or breathlessness.
Pulmonary fibrosis can occur in advanced RA and may cause dyspnoea . The risk of atheroma and cardiovascular
disease is increased in RA.
This is caused by a combination of conventional risk factors, such as high cholesterol, smoking, hypertension and reduced physical activity, and the effects of inflammatory factors, such as immune complexes, cytokines and chemokines, on the endothelium and adipose tissue.
The risk of cardiovascular disease may be increased by NSAID and corticosteroids, whereas there is some evidence
that DMARD and biological medicines are protective.
Neurological complications
Peripheral entrapment neuropathies may result fromcompression by hypertrophied synovium or by joint
subluxation. Median nerve compression in the carpal
tunnel is most common and bilateral compression can
occur as an early presenting feature of RA. Other syndromes include ulnar nerve compression at the elbow or wrist, compression of the lateral popliteal nerve at the
head of the fibula, and tarsal tunnel syndrome (entrapment of the posterior tibial nerve in the flexor retinaculum),which causes burning, tingling and numbness in
the distal sole and toes. Diffuse symmetrical peripheral
neuropathy and mononeuritis multiplex may occur in
patients with rheumatoid vasculitis.
Cervical cord compression can result from subluxation
of the cervical spine at the atlanto-axial joint or at a
subaxial level . Atlanto-axial subluxation is a
common finding in long-standing RA and is due to
erosion of the transverse ligament posterior to the
odontoid peg. If unrecognised, it can lead to cord
compression or sudden death following minor trauma
or manipulation. Atlanto-axial subluxation should be
suspected in any RA patient who describes new onset of
occipital headache, particularly if symptoms of paraesthesia or electric shock are present in the arms. The onset is often insidious, with subtle loss of function that is
initially attributed to active disease.
Reflexes and power can be very difficult to assess in the presence of marked joint damage and therefore sensory or upper motor signs are most important. Patients with spinal cord compression may require operative stabilisation and fixation, though the outcome is poor if the patient already has tetraparesis.
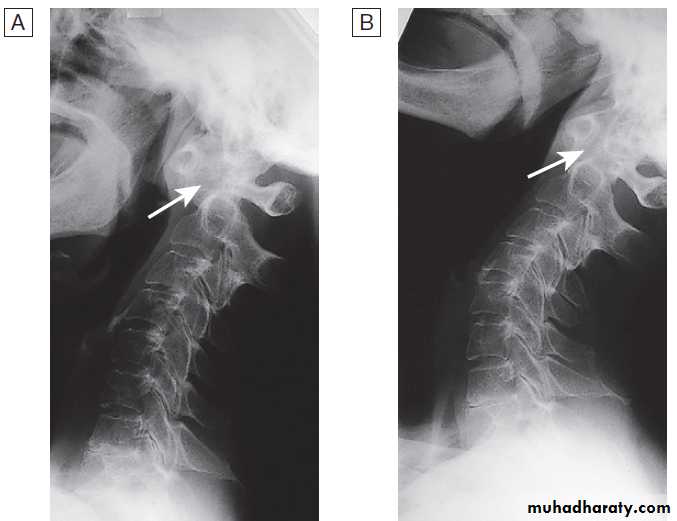
Subluxation of cervical spine.
A Flexion, showingwidening of the space (arrow) between the odontoid peg of the axis (behind) and the anterior arch of the atlas (in front).
B Extension, showing
reduction in this space.
Other complications
Amyloidosis is a rare complication of prolonged active
disease and usually presents with nephrotic syndrome.
Microcytic anaemia can occur due to iron deficiency
resulting from NSAID-induced gastrointestinal blood loss, and normochromic, normocytic anaemia with thrombocytosis is present in active disease. Felty’s syndrome is the association of splenomegaly and neutropenia with RA . Generalised and local lymphadenopathy affecting nodes draining actively inflamed joints may both occur. Patients with persistent lymphadenopathy should be biopsied since there is an increased risk of lymphoma with longstanding RA.
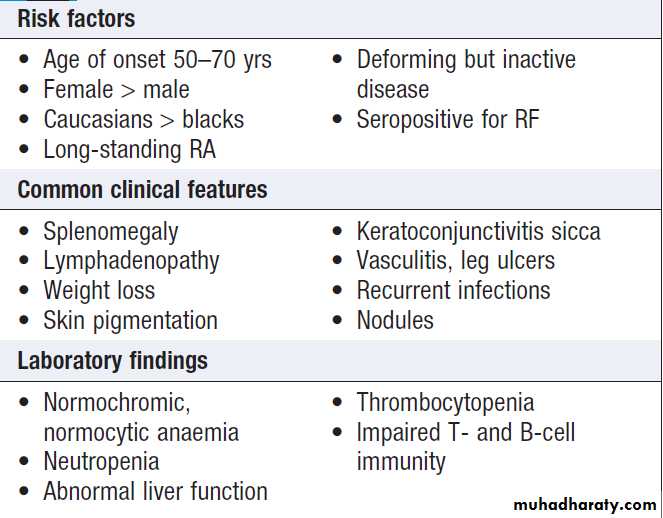
Felty’s syndrome
InvestigationsThe diagnosis of RA is based on clinical grounds but
investigations are useful in confirming the diagnosis and
assessing disease activity .The ESR and CRP
are usually raised but normal results do not exclude the
diagnosis. ACPA are positive in about 70% of cases and
are highly specific for RA, occurring in many patients
before clinical onset of the disease. Similarly RF is positive
in about 70% of cases, many of whom also test positive
for ACPA. Low titres of RF are found in about 10%
of the normal population and in other diseases .
Ultrasound examination and MRI are not routinely
required in patients with obvious clinical signs.
Their main value is in patients with symptoms suggestive of
an inflammatory arthritis, where there is uncertainty
about the presence of synovitis.
They are also a sensitive means of detecting early erosions. Plain X-rays of the hands, wrist and feet are of limited value in early RA but certain changes are characteristic, including periarticular osteoporosis and marginal joint erosions.
The main indication for X-ray is in the assessment of patients with problem joints to determine if significant structural damage has occurred.Patients who are suspected of having atlanto-axial disease should have lateral X-rays taken in flexion and extension, and the degree of cord compression should be established with MRI.
In patients with Baker’s cyst, ultrasound may be required
to establish the diagnosis, since DVT and Baker’s cystmay coexist.
The DAS28 score is widely used to assess disease
activity, the response to treatment and the need for
biological therapy. It involves counting the number
of swollen and tender joints in the upper limbs
and knees, and combining this with the ESR and the
patient’s assessment of his/her general health on a visual analogue scale, to generate a numerical score. The
higher the value, the more active the disease .
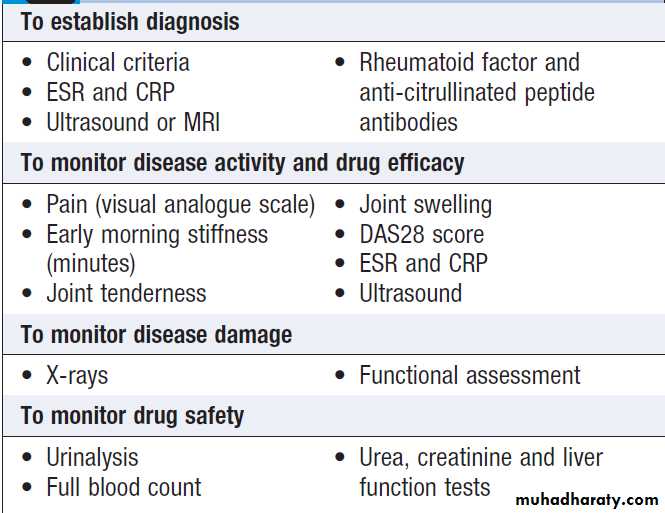
Investigations and monitoring of rheumatoid arthritis
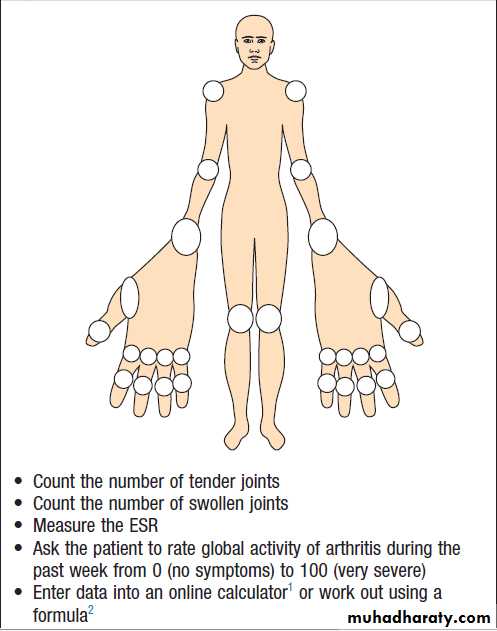
How to calculate the DAS28 score
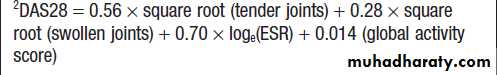
Management
The mainstay of treatment in RA comprises the earlyuse of small-molecule disease-modifying antirheumatic
drugs (DMARDs), and corticosteroids for induction of remission. There is evidence that early use of DMARD
therapy improves clinical outcome in RA. Partial or nonresponse to DMARD therapy should prompt escalation
of the dose or use of an additional DMARD, with progression to biological drugs if necessary .
Regular monitoring of DMARD therapy is essential
because of the risk of liver and haematological toxicity.
Some DMARDs are contraindicated in pregnancy, especially during the first trimester
Patients who wish to become pregnant should be counselled to stop DMARD treatment while they try to conceive. Since RA almost always undergoes remission during pregnancy, it is usually possible to manage the condition without DMARD therapy over this period.

Management of early rheumatoid arthritis.
(DMARD = disease-modifying antirheumatic drug; TNF = tumour necrosis factor)
Rheumatoid arthritis in pregnancy
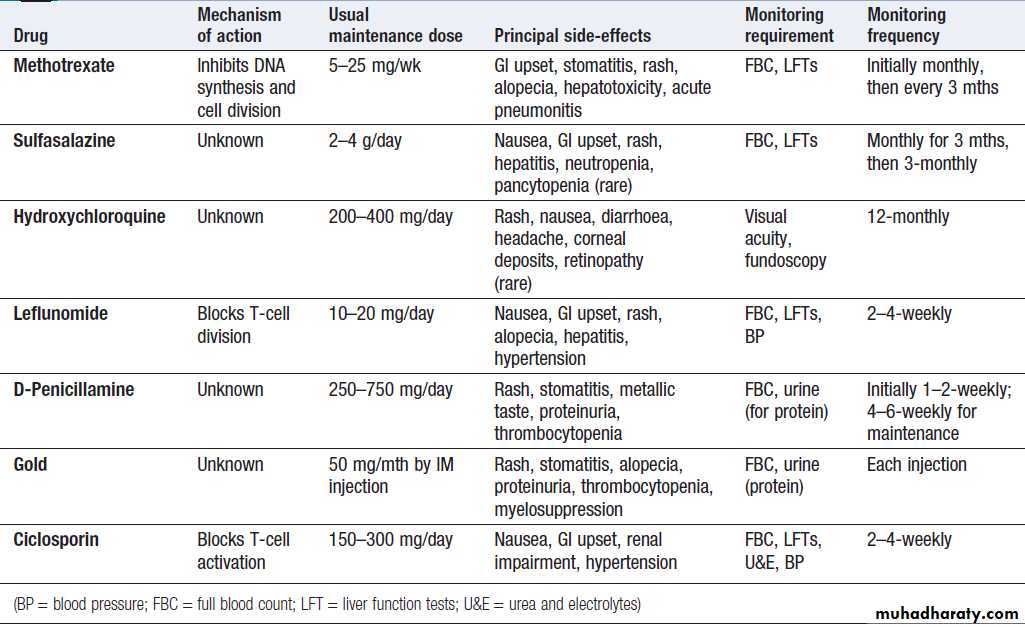
Commonly used small-molecule disease-modifying antirheumatic drugs in rheumatoid arthritis
Disease-modifying antirheumatic drugsMethotrexate
Methotrexate is the anchor DMARD in RA. It is usually
given as a starting weekly oral dose of 7.5–10 mg and
this is increased in 2.5 mg increments every 2–4 weeks
until benefit occurs or toxicity is limiting. The maximum
recommended dose is 25 mg. The benefits usually start to appear within 1–2 months but a 6-month course should be given before concluding that it has been ineffective. The most common adverse effects are nausea, vomiting and malaise within 24–48 hours of administration. Patients who experience these can sometimes be successfully treated with subcutaneous methotrexate.Folic acid (5 mg/week) reduces the incidence of adverse effects without reducing efficacy.
Patients should be warned of drug interaction with sulphonamides and to avoid excess alcohol, which enhances methotrexate hepatotoxicity. Acute pulmonary toxicity (pneumonitis) is rare but can occur at any time. Patients should therefore be warned to seek early advice if they develop any new respiratory symptoms. Methotrexate should be stopped immediately if pneumonitis is suspected and high-dose steroids given.
Sulfasalazine (SSZ)
Is widely used, both alone and in combination with methotrexate and other drugs. The mechanism of action is incompletely understood. Nausea and gastrointestinal intolerance are the main adverse effects.
The usual starting dose is 500 mg daily, building up gradually to a maintenance dose of 2–4 g/day. The patient should be warned of possible orange staining of urine and contact lenses. Monitoring should be performed for liver and haematological toxicity.
Hydroxychloroquine
Hydroxycholoroquine is given in a dose of 400 mg daily,
usually in combination with other DMARDs. Ocular toxicity can occur with long-term use due to retinal damage. It is usual to check visual acuity before starting treatment and to repeat this periodically whilst treatment is continued.
Leflunomide
can be used alone or in combination. It works by inhibiting lymphocyte proliferation and activation. It is usually well tolerated and has low marrow toxicity, but may cause liver dysfunction. The usual maintenance dose is 10–20 mg/day. It is possible to give a loading dose of 100 mg on 3 consecutive days at the start of treatment, but this is seldom used in routine clinical practice. Monitoring should be performed for liver and haematological toxicity.Gold, penicillamine and ciclosporin A
These are only occasionally used due to the availabilityof drugs with a better risk–benefit profile. Gold (sodium
aurothiomalate) is given by deep intramuscular injection
of 50 mg after an initial 10 mg test dose. Treatment is
continued weekly for up to 6 months until benefit occurs,
when the frequency is reduced to fortnightly and then monthly. Penicillamine is given in a starting dose of
125–250 mg daily on an empty stomach, and increased
in 125-mg increments every 6 weeks until benefit occurs
or adverse effects develop.
Ciclosporin A inhibits lymphocyte division and activation. It is given in a dose of 150–300 mg/day with monitoring of blood pressure and renal function.
Corticosteroids
Systemic corticosteroids have disease-modifying activity,but their primary role is in the induction of remission
in patients with early RA who are starting synthetic
DMARD treatment. Various regimens have been used
but there is little evidence to suggest that one is superior
to another. One strategy is to give a high dose of
oral prednisolone initially (60 mg daily) and to reduce
and stop this gradually over a period of 3 months as the
DMARD starts to take effect. Another is to employ low-dose prednisolone (5–10 mg daily for 6–24 months) or
to give intramuscular injections of methylprednisolone
or triamcinolone every 6–8 weeks. Intramuscular steroids
are often used to treat flares of disease activity in
patients who are established on DMARD therapy.
Intraarticular corticosteroids are primarily indicated when
there are one or two ‘problem joints’ with persistent
synovitis despite good general control of the disease.
Although corticosteroids are very useful, they also
have significant adverse effects . In the context
of RA, osteoporosis is probably the most important
since this is a known complication of RA, even in the
absence of corticosteroid therapy.
Accordingly DEXA scanning followed by bone protection should be considered in any patient with RA who is expected to be on more than 7.5 mg prednisolone daily for more than 3 months.
Biological therapies
The use of biological agents (often abbreviated to ‘biologics’) is reserved for the treatment of patients whohave high disease activity despite having had an adequate trial of traditional DMARDs. These agents are
targeted towards specific cytokines and other cell-surface
molecules regulating the immune response.
Although generally well tolerated, biological therapies
increase the risk of serious infections due to suppression
of the immune response. Although biological treatment
is more effective than standard DMARD therapy, treatment
costs are significantly greater.
Because of this, many countries have set guidelines restricting their use.
Current UK recommendations are that biological therapy
should be initiated only in active RA (DAS28 > 5.1;
p. 1100) when an adequate trial of at least two other
DMARDs (including methotrexate) has failed. Details of
the individual agents, their mechanisms of action and
their toxicity are shown in Box.
Anti-TNF therapy
Is the first-line biological drug in RA. Several agents are available, as summarised in Box. With the exception of infliximab, which must be prescribed with methotrexate to reduce the risk of neutralizing antibodies developing, these agents can be used as monotherapy. In clinical practice, however, most are co-prescribed with methotrexate, as this is more efficacious. The main adverse effects are serious infections and reactivation of latent tuberculosis. There is evidence that TNF blockade can increase the risk of some malignancies, particularly basal cell carcinoma of the skin, and that it can accelerate progression of cancer in patients with prior malignant disease. In contrast, the risk of vascular disease in RA patients seems to be reduced by anti-TNF therapy.Rituximab
Rituximab is an antibody directed against the CD20receptor, which is expressed on B lymphocytes and
immature plasma cells. It is given by two intravenous
infusions, 2 weeks apart, usually in combination with
intravenous corticosteroid. It causes depletion of peripheral and synovial B cells, which is sustained for several months after administration.
The treatment is repeated usually when signs of improvement are wearing off (anything from 6 months to 1 year). It is mostly used in the treatment of patients with resistant RA who have failed to respond to TNF blockade.
Abatacept
Abatacept is an agent in which the Fc domain of IgG isfused to the extracellular domain of CTLA4. It blocks the
interaction between CD28 and CD80/86 that is required
for full activation of T cells following antigen presentation
by dendritic cells or macrophages. Abatacept has a
good safety profile and is as efficacious as anti-TNF
therapy in biological-naïve patients who fail to respond
to conventional DMARD therapy.
Tocilizumab
This agent is an antibody directed against the IL-6 receptor, and is licensed for the treatment of RA . It prevents IL-6 activating its receptor within the synovial membrane and in other tissues such as liver and muscle.
It has similar efficacy to anti-TNF therapy and is licensed for use as monotherapy or in combination with methotrexate. Adverse effects include leucopenia,
hypercholesterolaemia and an increased risk of infections. It is generally used as second-line treatment in patients who have failed to respond to anti-TNF therapy, except in those who are intolerant of methotrexate, in which case it is used as a first-line treatment.
Anakinra
Anakinra is a decoy receptor for IL-1. It has some activity
in RA but is seldom used since it appears to be less
effective than other biological drugs.
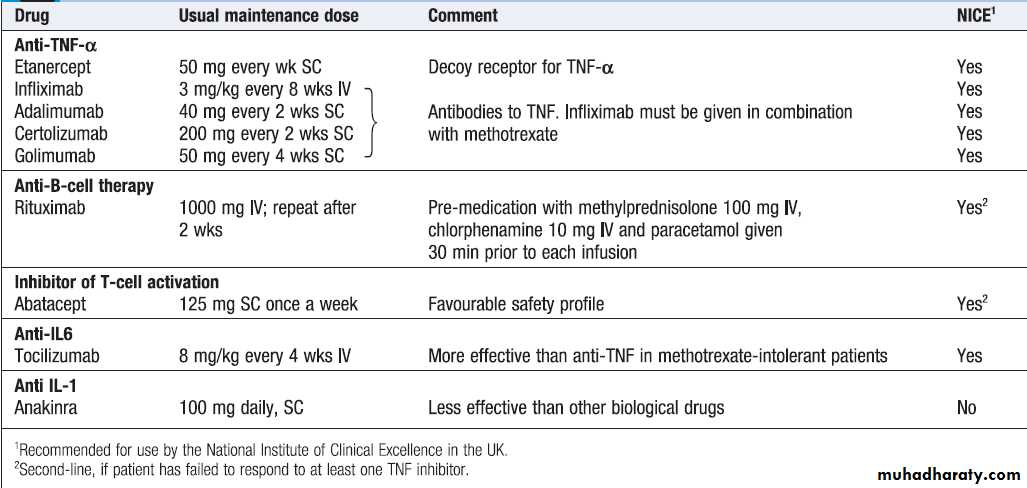
Commonly used biological drugs in rheumatoid arthritis
Other treatmentsSurgery
Synovectomy of the wrist or finger tendon sheaths of the
hands may be required for pain relief or to prevent
tendon rupture when medical interventions have failed.
In later stages when joint damage has occurred, osteotomy, arthrodesis or arthroplasty may be required . General measures
The general principles should be followed.
Physical rest, analgesics and NSAID may be required to control symptoms. Passive exercises and joint protection measures should be encouraged with the aim of conserving function in affected joints.
During treatment, periodic assessment of disease activity, progression and disability is essential.
In the vast majority, management is outpatient- or day patient-based, but hospital admission can be helpful in patients with very active disease for a period of bed rest, multiple joint injections, splinting, regular hydrotherapy, physiotherapy and education.
JUVENILE IDIOPATHIC ARTHRITIS
Inflammatory arthritis occurs rarely in children. Severaldistinct subtypes are recognised (Box). Systemic
juvenile idiopathic arthritis (JIA; formerly known as Still’s disease) is a systemic disorder characterised by
fever, rash, arthritis, hepatosplenomegaly and serositis
in association with a raised ESR and CRP. Autoantibody
tests are negative. Oligoarthritis is the most common form, accounting for about 60% of cases. Two subtypes are recognised, depending on the extent of involvement, but they probably form part of a single disease spectrum. Oligoarticular JIA is more common in females and tends to affect large joints in an asymmetrical pattern.
There is an association with uveitis and many patients are ANA-positive.
Rheumatoid factor-negative polyarthritis is a heterogeneous form. Some patients are ANA-positive andthese probably represent a subtype of oligoarticular JIA
with more extensive joint involvement. Other patients
present with polyarticular disease that is similar to adult
seronegative RA. Juvenile forms of ankylosing spondylitis
and psoriatic arthritis also occur. Patients with JIA should be referred to a paediatric rheumatologist. The principles of management are similar to those in adult inflammatory disease, and corticosteroids and methotrexate are required for systemic JIA.
Recent trials indicate that TNF blockers, IL-1 inhibitors
and tocilizumab may also be effective. Steroids andmethotrexate are also indicated for oligoarticular and
polyarticular JIA, with progression to anti-TNF therapy
in poor responders.
The prognosis of oligoarthritis is good and in many
patients the condition resolves at puberty. Polyarticular
and systemic JIA have a poorer prognosis, however, and
in about 50% of cases the disease remains active into
adulthood, requiring extended follow-up by adult rheumatology services.
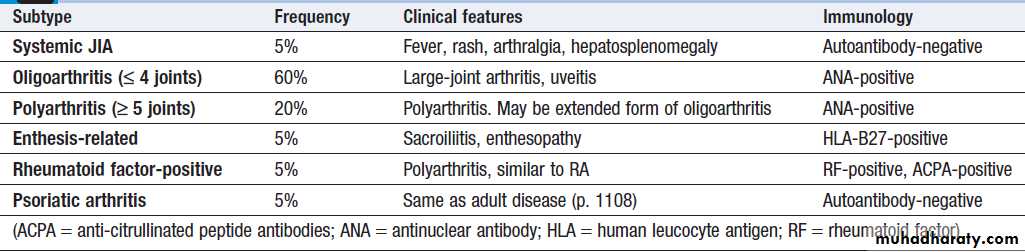
Clinical features of juvenile idiopathic arthritis

Juvenile idiopathic arthritis in adolescence
Adult-onset Still’s diseaseThis is a rare systemic inflammatory disorder of unknown cause, similar to juvenile idiopathic arthritis, which presents with intermittent fever, rash and arthralgia. Splenomegaly, hepatomegaly and lymphadenopathy may be present. Investigations typically show evidence of an acute phase response, with a markedly elevated serum ferritin. Tests for RF and ANA are negative. Most patients respond to corticosteroids but DMARDs may also be required as steroid-sparing agents. Anecdotal reports indicate that anakinra , anti-TNF therapy and tocilizumab may be helpful in patients with resistant disease, but none
of these has been tested in a randomised trial.
SERONEGATIVE SPONDYLOARTHROPATHIES
These comprise a group of related inflammatory jointdiseases, which show considerable overlap in their clinical
features and a shared immunogenetic association
with the HLA-B27 antigen . They include:
• ankylosing spondylitis
• axial spondyloarthritis
• reactive arthritis, including Reiter’s syndrome
• psoriatic arthritis
• arthropathy associated with inflammatory bowel disease.
The synovitis is non-specific and is often indistinguishable
from RA. However, the distinctive feature of this group of diseases is the marked degree of extrasynovial inflammation, especially of the enthesis but also of the joint capsule, periosteum, cartilage and subchondral bone.
There is a striking association with carriage of the HLA-B27 allele, particularly for ankylosing spondylitis (> 95%) and reactive arthritis (90%), and especially associated with sacroiliitis, uveitis or balanitis. Understanding of the cause is incomplete but an aberrant response to infection is thought to be involved in genetically predisposed individuals. In some situations, a triggering organism can be identified, as in reactive arthritis following bacterial dysentery or chlamydial urethritis, but in others the environmental trigger remains obscure. Familial clustering not only is common to the specific condition occurring in the proband, but also may extend to other seronegative spondyloarthopathy group.
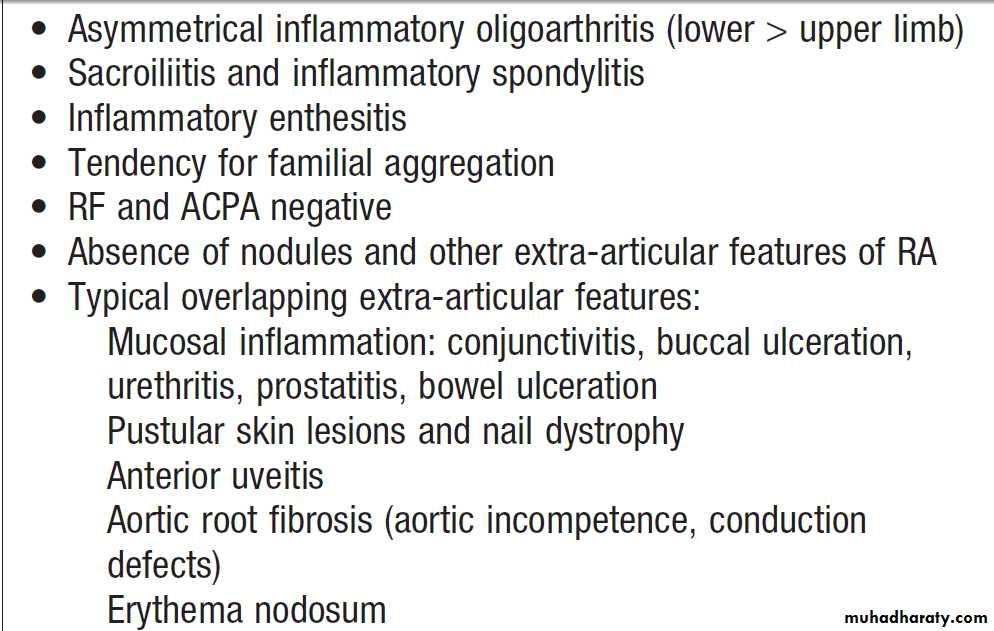
Clinical features common to seronegative
spondyloarthritisAnkylosing spondylitis
Ankylosing spondylitis (AS) is characterised by a chronicinflammatory arthritis predominantly affecting the
Sacroiliac joints and spine, which can progress to bony
fusion of the spine. The onset is typically between the
ages of 20 and 30, with a male preponderance of about
3 : 1. In Europe, more than 90% of those affected are
HLA-B27-positive.
The overall prevalence is less than 0.5% in most populations. Over 75% of patients are able to remain in employment and enjoy a good quality of life.
Even if severe ankylosis develops, functional limitation
may not be marked as long as the spine is fused in
an erect posture.
Pathophysiology
Ankylosing spondylitis is thought to arise from an as yet
ill-defined interaction between environmental pathogens
and the host immune system in genetically susceptible
individuals. Increased faecal carriage of Klebsiella
aerogenes may relate to exacerbation of both joint and eye disease. Wider alterations in the human gut microbial environment are increasingly implicated, which could lead to increased levels of circulating cytokines. The HLA-B27 molecule is implicated through its antigen-presenting function (it is a class I MHC molecule) or activate leucocytes. This could lead to inflammatory cytokine release by macrophages and dendritic cells, thus triggering inflammatory disease.
Clinical features
The cardinal feature is low back pain and early morningstiffness with radiation to the buttocks or posterior
thighs. Symptoms are exacerbated by inactivity and
relieved by movement. The disease tends to ascend
slowly, ultimately involving the whole spine, although
some patients present with symptoms of the thoracic
or cervical spine.
As the disease progresses, the spine becomes increasingly rigid as ankylosis occurs.
Secondary osteoporosis of the vertebral bodies frequently occurs, leading to an increased risk of vertebral fracture. Early physical signs include a reduced range of lumbar spine movements in all directions and pain on sacroiliac stressing.
As the disease progresses, stiffness increases throughout the spine and chest expansion becomes restricted.
Spinal fusion varies in its extent and in most cases does not cause a gross flexion deformity, but a few patients develop marked kyphosis of the dorsal and cervical spine that may interfere with forward vision. This may prove incapacitating, especially when associated with fixed flexion contractures of hips or knees. Pleuritic chest pain aggravated by breathing is common and results from costovertebral joint involvement.
Plantar fasciitis, Achilles tendinitis and tenderness
over bony prominences such as the iliac crest and greater
trochanter may all occur, reflecting inflammation at the
sites of tendon insertions (enthesitis).
Up to 40% of patients also have peripheral arthritis.
This is usually asymmetrical, affecting large joints such
as the hips, knees, ankles and shoulders. In about 10%
of cases, involvement of a peripheral joint may antedate
spinal symptoms, and in a further 10%, symptoms begin
in childhood, as in the syndrome of oligoarticular juvenile
idiopathic arthritis.
Fatigue is a major complaint and may result from
both chronic interruption of sleep due to pain, and
chronic systemic inflammation with direct effects of
inflammatory cytokines on the brain.
Acute anterior uveitis is the most common extra-articular feature, which occasionally precedes joint disease. Other extraarticular features are occasionally observed but are rare . Disease activity in AS can be assessed by the
Bath Ankylosing Spondylitis Disease Activity Index
(BASDAI), a questionnaire in which patients and their
physician rate severity of various symptoms .
This is important in assessing eligibility for biological
treatment. The criteria for the diagnosis of classical AS
and axial spondyloarthropathy (SpA) are shown in Box .
The criteria for axial SpA were developed to take account of the fact that X-rays may be normal in early AS.

Extra-articular features of ankylosing spondylitis
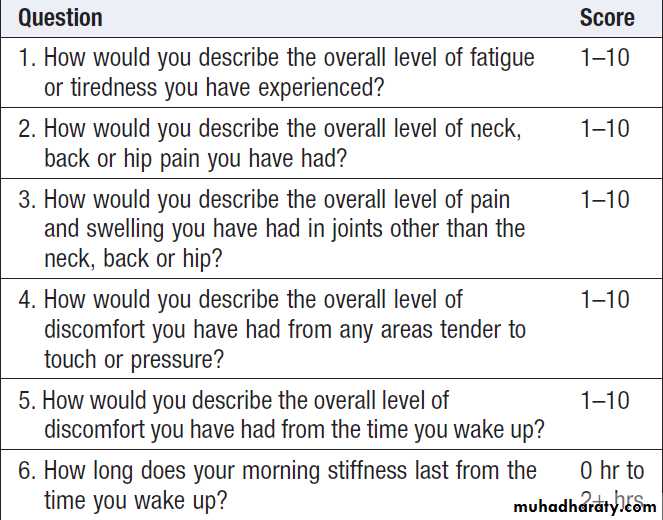
The patient is asked to complete each question. The score is
calculated by taking the average of all 6 questions, where duration of morning stiffness in minutes is coded in 12-minute increments from none (1) to 120 (10).Bath Ankylosing Spondylitis Disease
Activity Index
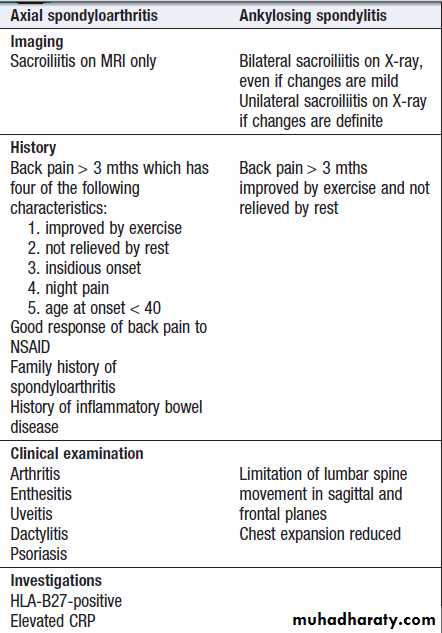
Axial spondyloarthritis is diagnosed when there is sacroiliitis
on MRI plus one other feature on history, clinical examinationor investigation. The diagnosis can also be made in patients
who are HLA-B27-positive with two or more clinical features
in the absence of sacroiliitis
Ankylosing spondylitis can be diagnosed when X-ray evidence
of sacroiliitis occurs with one other feature on history or
clinical examination
Diagnostic criteria for ankylosing spondylitis and axial spondyloarthritis
Investigations
In established AS, radiographs of the sacroiliac joint
show irregularity and loss of cortical margins, widening
of the joint space and subsequently sclerosis, joint space
narrowing and fusion. Lateral thoracolumbar spine
X-rays may show anterior ‘squaring’ of vertebrae due to
erosion and sclerosis of the anterior corners and periostitis of the waist.
Bridging syndesmophytes may also be seen. These are areas of calcification that follow the outermost
fibres of the annulus . In advanced disease, ossification of the anterior longitudinal ligament and facet joint fusion may also be visible. The combination of these features may result in the typical ‘bamboo’ spine .
Erosive changes may be seen in the symphysis pubis, the ischial tuberosities and peripheral joints. Osteoporosis and atlanto-axial dislocation can occur as late features. Patients with early disease can have normal X-rays, and if clinical suspicion is high, MRI should be performed. This is much more sensitive for detection of early sacroiliitis than X-ray and can also detect inflammatory changes in the lumbar spine. The ESR and CRP are usually raised in active disease but may be normal. Testing for HLA-B27 can be helpful, especially in patients with back pain suggestive of an inflammatory cause, when other investigations have yielded equivocal results.
Autoantibodies such as RF, ACPA and ANA are negative.
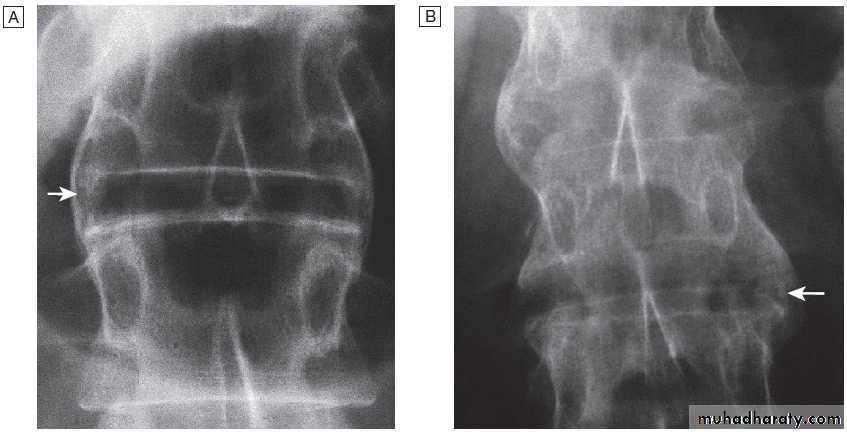
X-ray changes in spondyloarthropathies.
A Fine symmetrical marginal syndesmophytes typical of ankylosing spondylitis (arrow).B Coarse, asymmetrical non-marginal syndesmophytes typical of psoriatic/Reiter’s spondylitis (arrow).
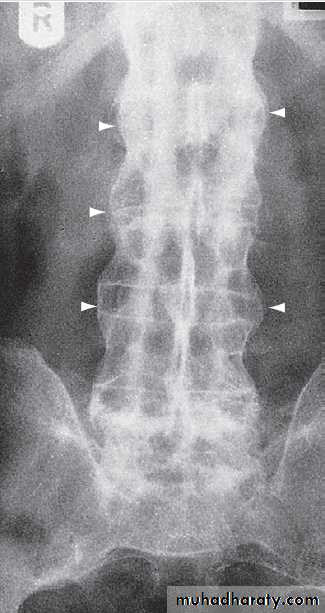
‘Bamboo’ spine of severe late ankylosing spondylitis.
Note the symmetrical marginal syndesmophytes (arrows), sacroiliac jointfusion and generalised osteopenia.
Management
The aims of management are to relieve pain and stiffness, maintain a maximal range of skeletal mobility and avoid the development of deformities. Education andappropriate physical activity are the cornerstones of
management. Early in the disease, patients should be
taught to perform daily back extension exercises, including a morning ‘warm-up’ routine, and to punctuate prolonged periods of inactivity with regular breaks.
Swimming is ideal exercise. Poor posture must be
avoided. NSAIDs and analgesics are often effective in
relieving symptoms and may alter the underlying course
of the disease. A long-acting NSAID at night is helpful
for alleviation of morning stiffness.
Peripheral arthritis can be treated with methotrexate or sulfasalazine, but these drugs have no effect on axial disease. Anti-TNF therapy should be considered in patients who are inadequately controlled on standard therapy with a BASDAI score of ≥ 4.0 and a spinal pain score of ≥ 4.0. Anti-TNF therapy frequently improves symptoms but has not been shown to prevent ankylosis or alter natural history of the disease.
Other biological interventions using agents developed for RA have been disappointing, suggesting fundamental differences in disease pathogenesis.
Local corticosteroid injections can be useful for persistent plantar fasciitis, other enthesopathies and peripheral arthritis.
Oral corticosteroids may be required for
acute uveitis but do not help spinal disease. Severe hip,
knee or shoulder restriction may require surgery. Total
hip arthroplasty has largely removed the need for difficult spinal surgery in those with advanced deformity.
Reactive arthritis
Reactive arthritis (previously known as Reiter’s disease)
is predominantly a disease of young men, with a male
preponderance of 15 : 1. It is the most common cause
of inflammatory arthritis in men aged 16–35 but may
occur at any age. Between 1 and 2% of patients with
non-specific urethritis seen at genitourinary medicine
clinics have reactive arthritis . Following an epidemic
of Shigella dysentery, 20% of HLA-B27-positive
men developed reactive arthritis. Reactive arthritis may
present with the triad described in Box but many
patients present with arthritis only.
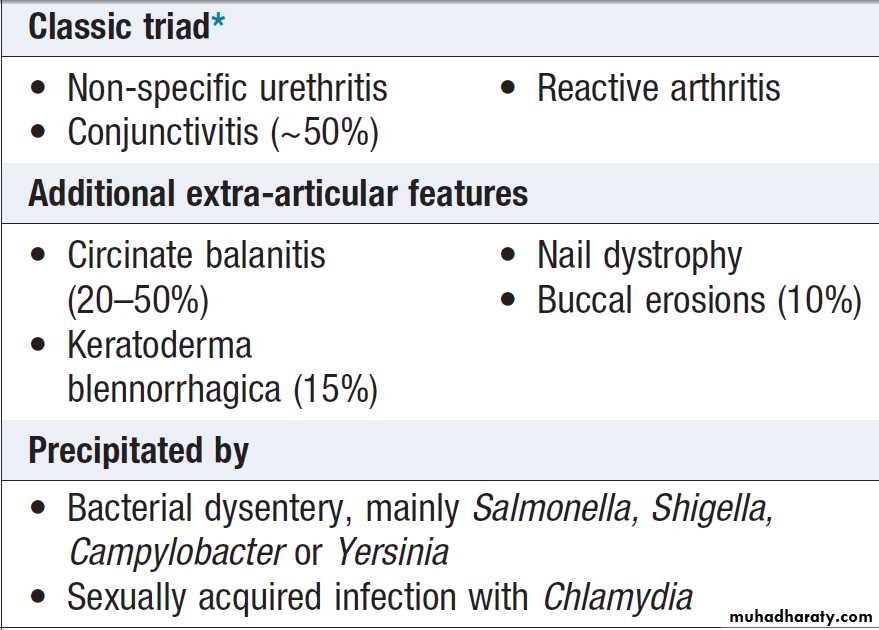
Reiter’s disease
Clinical featuresThe onset is typically acute, with an inflammatory
Oligoarthritis that is asymmetrical and targets lower
limb joints, such as the knees, ankles, midtarsal and
MTP joints. It occasionally presents with single joint involvement and no clear history of an infectious trigger.
There may be considerable systemic disturbance, with
fever and weight loss. Achilles tendinitis or plantar fasciitis may also be present.
The first attack of arthritis is usually self-limiting, but recurrent or chronic arthritis develops in more than 60% of patients, and about 10% still have active disease 20 years after the initial presentation.
Low back pain and stiffness are common and 15–20% of patients develop sacroiliitis. Spondylitis, chronic erosive arthritis, recurrent acute arthritis and uveitis are the major causes of long-term morbidity.
Several extra-articular features may occur (Box). Circinate balanitis starts as vesicles on the coronal margin of the prepuce and glans penis, later rupturing to form superficial erosions with minimal surrounding erythema, some coalescing to give a circular pattern. Lesions are often painless and may escape notice.
Keratoderma blennorrhagica begins as discrete waxy, yellow-brown vesico-papules with desquamating margins, coalescing to form large crusty plaques.
The palms and soles are particularly affected
but spread may occur to the scrotum, scalp and trunk.These lesions are indistinguishable from pustular psoriasis.
Nail dystrophy with subungual hyperkeratosis is
common and indistinguishable from psoriatic nail dystrophy.
Mouth ulcers manifest as shallow red painless
patches on tongue, palate, buccal mucosa and lips,
lasting only a few days. Conjunctivitis may accompany
the first acute episode. Uveitis is rare with the first attack
but occurs in 30% of patients with recurring or chronic
arthritis. Rare complications include aortic incompetence, conduction defects, pleuropericarditis, peripheral neuropathy, seizures and meningoencephalitis.
Investigations
The diagnosis is usually made clinically but joint aspirationmay be required to exclude crystal arthritis and
articular infection. Synovial fluid is leucocyte-rich and
may contain multinucleated macrophages (Reiter’s cells). ESR and CRP are raised during an acute attack.
Urethritis may be confirmed in the ‘two-glass test’ by
demonstration of mucoid threads in the first-void specimen
that clear in the second. High vaginal swabs may
reveal Chlamydia on culture.
Except for post-Salmonella arthritis, stool cultures are usually negative by the time the arthritis presents, but serum agglutinin tests may help confirm previous dysentery. RF, ACPA and ANA are negative.
In chronic or recurrent disease, X-rays show periarticular osteoporosis, joint space narrowing and
proliferative erosions. Another characteristic feature is
periostitis, especially of metatarsals, phalanges and
pelvis, and large, ‘fluffy’ calcaneal spurs. In contrast to
AS, radiographic sacroiliitis is often asymmetrical and
sometimes unilateral, and syndesmophytes are predominantly coarse and asymmetrical, often extending
beyond the contours of the annulus (‘non-marginal’).
X-ray changes in the peripheral joints and spine are identical to those in psoriasis.
Management
The acute attack should be treated with rest, oral NSAIDsand analgesics. Intra-articular steroids may be required in patients with severe synovitis. Non-specific chlamydial
urethritis is usually treated with a short course of
doxycycline or a single dose of azithromycin, and this
may reduce the frequency of arthritis in sexually
acquired cases. Treatment with DMARDs should be considered for patients with persistent marked symptoms,
recurrent arthritis or severe keratoderma blennorrhagica.
Anterior uveitis is a medical emergency requiring
topical, subconjunctival or systemic corticosteroids.
Psoriatic arthritis
Psoriatic arthritis (PsA) occurs in 7–20% of patients withpsoriasis and in up to 0.6% of the general population.
The onset is usually between 25 and 40 years of age.
Most patients (70%) have pre-existing psoriasis but
in 20% the arthritis predates the occurrence of skin
disease. Occasionally, the arthritis and psoriasis develop
synchronously.
Clinical features
The presentation is with pain and swelling affecting the
joints and entheses. Several patterns of joint involvement
are recognised but the course is generally one of
intermittent exacerbation followed by varying periods
of complete or near-complete remission .
Destructive arthritis and disability are uncommon, except in the case of arthritis mutilans.
• Asymmetrical inflammatory oligoarthritis affects about
40% of patients and often presents abruptly with a
combination of synovitis and adjacent periarticular
inflammation. This occurs most characteristically in
the hands and feet, when synovitis of a finger or toe
is coupled with tenosynovitis, enthesitis and
inflammation of intervening tissue to give a ‘sausage digit’ or dactylitis . Large joints, such as the knee and ankle, may also be involved, sometimes with very large effusions.
• Symmetrical polyarthritis occurs in about 25% of
cases. It predominates in women and may stronglyresemble RA, with symmetrical involvement of
small and large joints in both upper and lower
limbs. Nodules and other extra-articular features
of RA are absent and arthritis is generally less
extensive and more benign. Much of the hand
deformity often results from tenosynovitis and soft
tissue contractures.
• Distal IPJ arthritis is an uncommon but characteristic
pattern affecting men more often than women. It targets finger DIP joints and surrounding periarticular tissues, almost invariably with accompanying nail dystrophy .
• Psoriatic spondylitis presents a similar clinical picture
to AS but with less severe involvement. It may
occur alone or with any of the other clinical patterns
described above and is typically unilateral or
asymmetric in severity.
• Arthritis mutilans is a deforming erosive arthritis
targeting the fingers and toes that occurs in 5%
of cases of PsA. Prominent cartilage and bone
destruction results in marked instability. The
encasing skin appears invaginated and ‘telescoped’
(‘main en lorgnette’) and the finger can be pulled
back to its original length.
Nail changes include pitting, onycholysis, subungual
hyperkeratosis and horizontal ridging. They are foundin 85% of those with PsA and only 30% of those with
uncomplicated psoriasis, and can occur in the absence
of skin disease. The characteristic rash of psoriasis
may be widespread, or confined to the scalp,
natal cleft and umbilicus, where it is easily overlooked.
Conjunctivitis can occur, whereas uveitis is mainly confined to HLA-B27-positive individuals with sacroiliitis
and spondylitis.
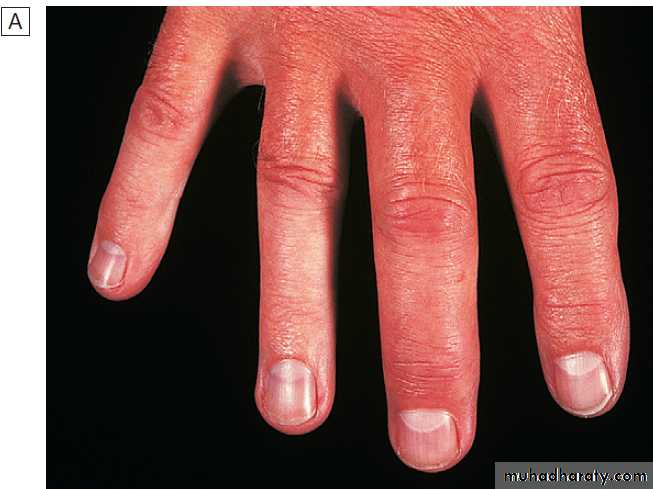
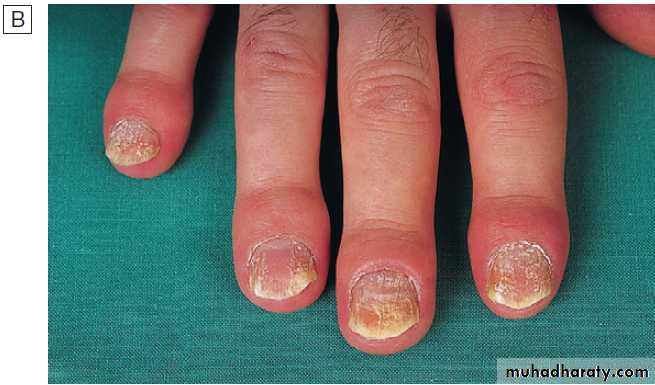
Psoriatic arthropathy.
A ‘Sausage’ middle finger of a patient with psoriatic arthritis.B Typical distal interphalangeal joint pattern with accompanying nail dystrophy (pitting and onycholysis).
Investigations
The diagnosis is made on clinical grounds. Autoantibodiesare generally negative and acute phase reactants,
such as ESR and CRP, are raised in only a proportion of
patients with active disease. X-rays may be normal or
show erosive change with joint space narrowing. Features
that favour PsA over RA include the characteristic
distribution of proliferative erosions with marked new bone formation, absence of periarticular osteoporosis and osteosclerosis. Imaging of the axial skeleton often reveals features similar to those in chronic reactive arthritis, with coarse, asymmetrical, non-marginal syndesmophytes and asymmetrical sacroiliitis. MRI and ultrasound with power Doppler are increasingly employed to detect synovial inflammation and inflammation at the entheses.
Management
Therapy with NSAID and analgesics may be sufficientto control symptoms in mild disease. Intra-articular
steroid injections can control synovitis in problem joints. Splints and prolonged rest should be avoided because
of the tendency to fibrous and bony ankylosis. Patients
with spondylitis should be prescribed the same exercise
and posture regime as in AS.
DMARDs should be considered for persistent synovitis unresponsive to conservative treatment. Methotrexate is the drug of first choice since it may also help skin psoriasis, but other DMARDs may also be effective, including sulfasalazine, ciclosporin and leflunomide.
DMARD monitoring should take place with particular attention to liver function since abnormalities are common in PsA. Hydroxychloroquine is generally avoided, as it can cause exfoliative skin reactions. Anti-TNF treatment should be considered for patients with active synovitis who respond inadequately to standard DMARDs.
This is effective for both PsA and psoriasis. Other biological treatments, such as ustekinumab,
are emerging, which target the IL-12/23 receptor.
Ustekinumab is highly effective in the treatment of psoriatic skin disease and is often effectve in PsA.
The retinoid acitretin is effective for skin lesions and, anecdotally, may also help arthritis, but it is teratogenic so must be avoided in young women.
It also causes mucocutaneous side-effects, hyperlipidaemia, myalgias and extraspinal calcification. Photochemotherapy with methoxypsoralen and long-wave ultraviolet light (psoralen + UVA, PUVA) is primarily used for skin disease, but can also help those with synchronous exacerbations of inflammatory arthritis.
Enteropathic arthritis
An acute inflammatory oligoarthritis occurs in around10% of patients with ulcerative colitis and 20% of those
with Crohn’s disease. It predominantly affects the large
lower limb joints (knees, ankles, hips) but wrists and
small joints of the hands and feet can also be involved.
The arthritis usually coincides with exacerbations of the
underlying bowel disease, and sometimes is accompanied
by aphthous mouth ulcers, iritis and erythema
nodosum. It improves with effective treatment of the
bowel disease, and can be cured by total colectomy in
patients with ulcerative colitis.
Patients with inflammatory bowel disease may also develop sacroiliitis (16%) and AS (6%), which are clinically and radiologically identical to classic AS. These can predate or follow the onset of bowel disease and there is no correlation between activity of the spondylitis and bowel disease.
The arthritis often remits with treatment of the bowel
disease but DMARD and biological treatment is occasionally required.
CONNECTIVE TISSUE DISEASES
These share overlapping clinical features, characterisedby dysregulation of immune responses, autoantibody
production often directed at components of the cell
nucleus, and widespread tissue damage.
Systemic lupus erythematosus (SLE)
Is a rare disease with a prevalence that ranges from about 0.03% in Caucasians to 0.2% in Afro-Caribbeans. Some 90% of affected patients are female and the peak age at onset is between 20 and 30 years. Lupus is associated with considerable morbidity and a five-fold increase in mortality compared to age- and gender-matched controls, mainly because of an increased risk of premature cardiovascular disease.
Pathophysiology
The cause of SLE is incompletely understood but geneticfactors play an important role. There is a higher concordance in monozygotic twins and the disease is strongly associated with polymorphic variants at the HLA locus. In a few instances, SLE is associated with inherited
mutations in complement components C1q, C2 and C4.
The characteristic feature is autoantibody production, which have specificity for a wide range of targets present within the cell or within the nucleus. This has led to the hypothesis that SLE may occur because of defects in apoptosis or in the clearance of apoptotic cells, which causes inappropriate exposure of intracellular antigens on the cell surface, leading to polyclonal B- and T-cell activation and autoantibody production.
Evironmental factors that cause flares of lupus, such as UV light and infections, increase oxidative stress and cause cell damage. Whatever the underlying cause of the autoantibody production, immune complex formation is thought to be an important mechanism of tissue damage in active SLE, leading to vasculitis and organ damage.
Clinical features
Symptoms such as fever, weight loss and mild lymphadenopathy may occur during flares of disease activity, whereas others such as fatigue, malaise and fibromyalgia like symptoms can be constant and not particularly associated with active inflammatory disease.
Arthritis
Arthralgia is a common symptom, occurring in 90% of
patients, and is often associated with early morning
stiffness. Tenosynovitis may also occur but clinically
apparent synovitis with joint swelling is rare. Joint
deformities may occur (Jaccoud’s arthropathy) as the
result of tendon damage, but joint erosions do not occur.
Raynaud’s phenomenon
Is common and may antedate other symptoms by months or years. A common presentation is Raynaud’s in combination with arthralgia or arthritis . Raynaud’s associated with
SLE and other connective tissue disease needs to be differentiated from primary Raynaud’s, which is common
in healthy young women.
Features in favour of secondary Raynaud’s include age at onset of over 25 years, absence of a family history of Raynaud’s, and occurrence in a male. Examination of capillary nail-fold loops using an ophthalmoscope (and oil placed on the skin) may help distinguish primary from secondary Raynaud’s. Loss of the normal loop pattern, with capillary ‘fallout’ and dilatation and branching of loops, supports connective tissue disease.
Skin
Rash is common in SLE and is classically precipitated by
exposure to UV light. Three distinct types occur:
• The classic butterfly facial rash (up to 20%). This is erythematous, raised and painful or itchy, and occurs over the cheeks with sparing of the nasolabial folds .
• The subacute cutaneous lupus erythematosus
(SCLE) rash, which is migratory, non-scarring andeither annular or psoriaform.
• The discoid lupus rash characterised by
hyperkeratosis and follicular plugging, with
scarring alopecia if it occurs on the scalp. Diffuse, usually non-scarring alopecia may also occur
with active disease. Other skin manifestations include
periungual erythema (reflecting dilated capillary loops), vasculitis and livedo reticularis , which is also a common feature of the antiphospholipid syndrome.
Severe secondary Raynaud’s leading to digital ulceration.
Butterfly (malar) rash of SLE, sparing the nasolabial folds.
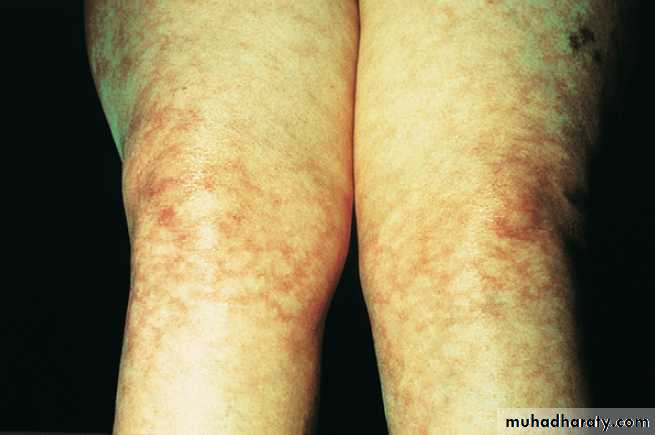
Livedo reticularis in SLE
KidneyRenal involvement is one of the main determinants of
prognosis, and regular monitoring of urinalysis and
blood pressure is essential. The typical renal lesion is a
proliferative glomerulonephritis , characterised
by heavy haematuria, proteinuria and casts on urine
microscopy.
Cardiovascular
The most common manifestation is pericarditis. Myocarditis
and Libman–Sacks endocarditis can also occur. The
endocarditis is due to accumulation on the heart valves
of sterile fibrin containing vegetations, which is thought
to be a manifestation of hypercoagulability associated
with antiphospholipid antibodies.
The risk of atherosclerosis is greatly increased, as is the risk of stroke and myocardial infarction. This is thought to be multifactorial due to the adverse effects of inflammation on the endothelium, chronic steroid therapy and the procoagulant effects of antiphospholipid antibodies.
Lung
Lung involvement is common and most frequentlymanifests as pleurisy or pleural effusion. Other features
include pneumonitis, atelectasis, reduced lung volume
and pulmonary fibrosis that leads to breathlessness. The
risk of thromboembolism is increased, especially in
patients with antiphospholipid antibodies.
Neurological
Fatigue, headache and poor concentration are common,
and often occur in the absence of laboratory evidence of
active disease. More specific features of cerebral lupus
include visual hallucinations, chorea, organic psychosis,
transverse myelitis and lymphocytic meningitis.
Haematological
Neutropenia, lymphopenia, thrombocytopenia orhaemolytic anaemia may occur, due to antibody-mediated destruction of peripheral blood cells. The
degree of lymphopenia is a good guide to disease
activity.
Gastrointestinal
Mouth ulcers may occur and may or may not be painful.
Mesenteric vasculitis is a serious complication, which
can present with abdominal pain, bowel infarction or
perforation.
Investigations
The diagnosis is based on a combination of clinical features and laboratory abnormalities. To fulfil the classification criteria for SLE, at least 4 of the 11 factors shown in Box must be present or have occurred in the
past. Patients should be screened for ANA and antibodies
to extractable nuclear antigens, and have complement
levels checked along with routine haematology and biochemistry. Patients with active SLE almost always test positive for ANA, but ANA-negative SLE can very rarely occur in the presence of antibodies to the Ro antigen. Anti-dsDNA antibodies are characteristic of severe active SLE but only occur in around 30% of cases.
Similarly, patients with active disease tend to have
low levels of C3 and C4, but this may be the result ofinherited complement deficiency that predisposes to
SLE. Studies of other family members can help to differentiate inherited deficiency from complement consumption.
A raised ESR, leucopenia and lymphopenia
are typical of active SLE, along with anaemia, haemolytic
anaemia and thrombocytopenia. CRP is often
normal in active SLE, except in the presence of serositis,
and an elevated CRP suggests coexisting infection.
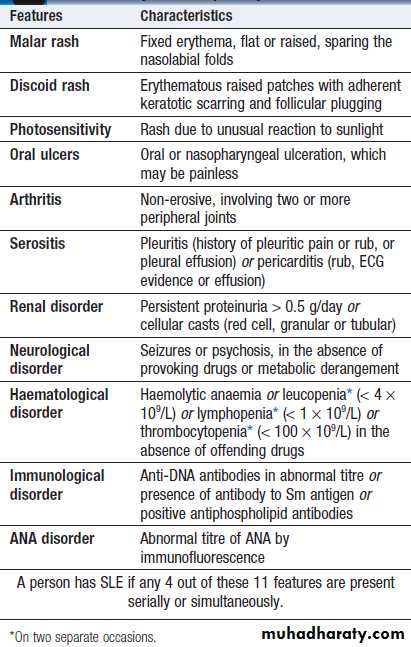
Revised American Rheumatism Association
criteria for systemic lupus erythematosusManagement
The therapeutic goals are to educate the patient about
the nature of the illness, to control symptoms and to prevent organ damage. Patients should be advised to
avoid sun and UV light exposure and to employ sun
blocks (factor 25–50).
Mild to moderate disease
Patients with mild disease restricted to skin and joints
can sometimes be managed with analgesics, NSAID
and hydroxychloroquine (200–400 mg daily). Frequently,
however, corticosteroids are also necessary (prednisolone 5–20 mg/day), often in combination with immunosuppressants such as methotrexate, azathioprine
or mycophenolate mofetil (MMF).
Increased doses of steroids may be required for flares in activity or complications such as pleurisy or pericarditis.
The monoclonal antibody belimumab, which targets the β-cell growth factor BLyS, has recently been shown to be effective in patients with active SLE who have responded inadequately to standard therapy.
Life-threatening disease
High-dose corticosteroids and immunosuppressants arerequired for the treatment of renal, CNS and cardiac
involvement. A commonly used regimen is pulse
Methylprednisolone (10 mg/kg IV), coupled with cyclophosphamide (15 mg/kg IV), repeated at 2–3-weekly intervals for six cycles. Cyclophosphamide may cause haemorrhagic cystitis, but the risk can be minimised
by good hydration and co-prescription of mesna,
which binds its urotoxic metabolites. Because of the
risk of azoospermia and anovulation (which may be permanent), pre-treatment sperm or ova collection and
storage need to be considered prior to treatment with
cyclophosphamide.
Mycophenolate mofetil has been used successfully in
combination with high-dose steroids for renal involvement
in SLE, with results equivalent to those of pulse
cyclophosphamide but fewer adverse effects. The role of
belimumab in life-threatening SLE remains to be established since clinical trials of this agent excluded patients with renal and cerebral lupus.
Maintenance therapy
Following control of the acute episode, the patient should be switched to oral immunosuppressive medication.
A typical regimen is to start oral prednisolone in a dose of 40–60 mg daily on cessation of pulse therapy, gradually reducing to reach a target of 10–15 mg/day or less by 3 months.
Azathioprine (2–2.5 mg/kg/day),
methotrexate (10–25 mg/week) or MMF (2–3 g/day)should also be prescribed. The long-term aim is to continue
the lowest dose of corticosteroids and immunosuppressant
that will maintain remission. Cardiovascular
risk factors, such as hypertension and hyperlipidaemia,
should be controlled and patients advised to stop
smoking.
Lupus patients with the antiphospholipid antibody
syndrome who have had previous thrombosis
require life-long warfarin therapy.
Systemic sclerosis
Systemic sclerosis, or scleroderma, is a generalised disorder of connective tissue affecting the skin, internal
organs and vasculature. It is characterised by sclerodactyly in combination with Raynaud’s and digital ischaemia (Fig.). The peak age of onset is in the
fourth and fifth decades, and overall prevalence is 10–20
per 100 000, with a 4 : 1 female preponderance. It is subdivided into diffuse cutaneous systemic sclerosis
(DCSS 30% of cases) and :
limited cutaneous systemic sclerosis
(LCSS: 70% of cases).
Many patients with LCSS have features that are phenotypically grouped into the ‘CREST’ syndrome (Calcinosis, Raynaud’s, oEsophageal involvement, Sclerodactyly and Telangiectasia). The
prognosis in DCSS is poor, with a 5-year survival of
approximately 70%. Features that associate with a poor
prognosis include older age, diffuse skin disease, proteinuria, high ESR, a low TLCO (gas transfer factor for
carbon monoxide) and pulmonary hypertension.
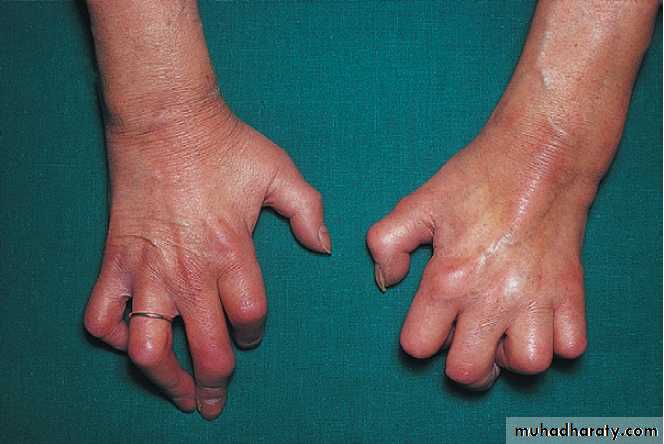
Systemic sclerosis. Hands showing tight, shiny skin,
sclerodactyly, flexion contractures of the fingers and thickening of the left middle finger extensor tendon sheath.Pathophysiology
The cause of systemic sclerosis is poorly understood.
There is evidence for a genetic component, and associations with alleles at the HLA locus have been found. The disease occurs in all ethnic groups, and race may influence severity, since DCSS is significantly more common
in black women than white. Isolated cases have been
reported in which a systemic sclerosis-like disease has
been triggered by exposure to silica dust, vinyl chloride,
hypoxyresins and trichloroethylene. There is clear evidence
of immunological dysfunction: T lymphocytes, especially those of the Th17 subtype, infiltrate the skin and there is abnormal fibroblast activation, leading to increased production of extracellular matrix in the dermis, primarily type I collagen.
This results in symmetrical thickening, tightening and induration of the skin (sclerodactyly). Arterial and arteriolar narrowing occurs due to intimal proliferation and vessel wall inflammation. Endothelial injury causes release of vasoconstrictors and platelet activation, resulting in further ischaemia, which is thought to exacerbate the fibrotic process.
Clinical features
SkinInitially, there is non-pitting oedema of fingers and
flexor tendon sheaths. Subsequently, the skin becomes
shiny and taut, and distal skin creases disappear. This is
accompanied by erythema and tortuous dilatation of
capillary loops in the nail-fold bed, readily visible with an ophthalmoscope .The face and neck are usually involved next, with thinning of the lips and radial furrowing. In some patients, skin thickening stops at this stage.
Skin involvement restricted to sites distal to the elbow or knee (apart from the face) is classified as ‘limited disease’or CREST syndrome .
Involvement proximal to the knee and elbow and on the trunk is classified as ‘diffuse disease’.
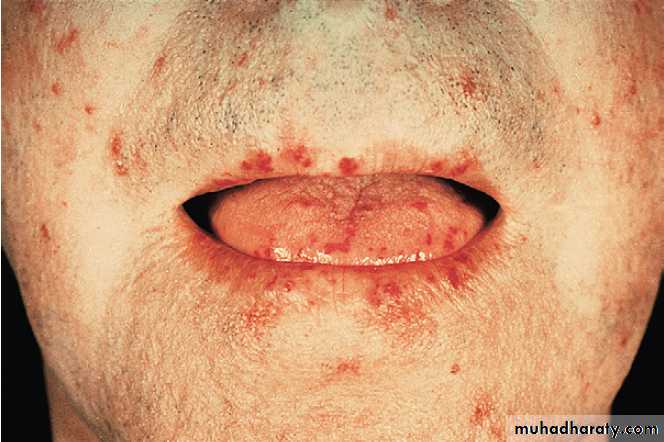
Typical facial appearance in the CREST syndrome.
Raynaud’s phenomenonThis is a universal feature and can precede other features
by many years. Involvement of small blood vessels in
the extremities may cause critical tissue ischaemia,
leading to skin ulceration over pressure areas, localised
areas of infarction and pulp atrophy at the fingertips.
Musculoskeletal features
Arthralgia, morning stiffness and flexor tenosynovitis
are common. Restricted hand function is due to skin
rather than joint disease and erosive arthropathy is
uncommon.
Muscle weakness and wasting can occur due to myositis.
Gastrointestinal involvement
Smooth muscle atrophy and fibrosis in the lower two-thirds of the oesophagus lead to reflux with erosive oesophagitis. Dysphagia and odynophagia may alsooccur. Involvement of the stomach causes early satiety
and occasionally outlet obstruction. Recurrent occult
upper gastrointestinal bleeding may indicate a water-melon’ stomach (antral vascular ectasia), which occurs in up to 20% of patients. Small intestine involvement
may lead to malabsorption due to bacterial overgrowth
and intermittent bloating, pain or constipation. Dilatation
of large or small bowel due to autonomic neuropathy
may cause pseudo-obstruction with nausea, vomiting, abdominal discomfort and distension, often worse after food.
Pulmonary involvement
This is a major cause of morbidity and mortality. Pulmonary hypertension complicates long-standing disease and is six times more prevalent in LCSS than in DCSS. It presents with rapidly progressive dyspnoea (more
rapid than interstitial lung disease), right heart failure
and angina, often in association with severe digital ischaemia. Fibrosing alveolitis mainly affects patients
with DCSS who have topoisomerase 1 antibodies.
Renal involvement
One of the main causes of death is hypertensive renal
crisis, characterised by rapidly developing malignant
hypertension and renal failure. Hypertensive renal crisis
is much more likely to occur in DCSS than in LCSS, and
in patients with topoisomerase 1 antibodies.
Investigations
Scleroderma is primarily a clinical diagnosis but variouslaboratory abnormalities are characteristic. The ESR is
usually elevated and raised levels of IgG are common,
but CRP values tend to be normal unless there is severe
organ involvement or coexisting infection. ANA is positive
in about 70%, and approximately 30% of patients
with DCSS have antibodies to topoisomerase 1 (Scl-70).
About 60% of patients with CREST syndrome have anticentromere antibodies.
Management
No treatments are available that halt or reverse the
fibrotic changes that underlie the disease. The focus of
management, therefore, is to ameliorate the effects of the
disease on target organs.
• Raynaud’s syndrome and digital ulcers. Raynaud’s
should be treated by avoidance of cold exposure
and use of mittens (heated mittens are available),
supplemented with calcium antagonists. Intermittent infusions of prostacyclin may benefit severe digital ischaemia. The endothelin 1 antagonist bosentan can be of value in promoting healing of digital ulcers. If these become infected, antibiotics may be required, but as these penetrate tissues poorly in scleroderma, they need to be given at higher doses for a longer duration than usual.
• Oesophageal reflux should be treated with proton
pump inhibitors and anti-reflux agents. Antibioticsmay be required for bacterial overgrowth
syndromes, and metoclopramide or domperidone
may help patients with symptoms of
pseudo-obstruction.
• Hypertension should be treated aggressively
with ACE inhibitors, even if renal impairment is
present.
• Joint involvement may be treated with analgesics
and/or NSAID. If synovitis is present, immunosuppressants such as methotrexate can also be of value.
• Pulmonary hypertension may be treated with
bosentan. In selected patients, heart–lung
transplantation may be considered. Corticosteroids
and cytotoxic drugs are indicated in patients who
have coexisting myositis or fibrosing alveolitis.
Mixed connective tissue disease
Mixed connective tissue disease (MCTD) is a conditionin which the clinical features of SLE, systemic sclerosis
and myositis may all occur in the same patient. It most
commonly presents with synovitis and oedema of the
hands, in combination with Raynaud’s phenomenon
and muscle pain or weakness.
Most patients have anti-ribonucleoprotein (RNP) antibodies, but these can occur in SLE without overlap features. Management focuses on treating the individual components of the syndrome.
Sjِgren’s syndrome
This is an autoimmune disorder of unknown cause,characterised by lymphocytic infiltration of salivary and
lachrymal glands, leading to glandular fibrosis and exocrine failure. The typical age of onset is between 40 and 50, with a 9 : 1 female preponderance. The disease may occur in isolation (primary Sjِgren’s syndrome) or in
patients with other autoimmune diseases (secondary
Sjِgren’s syndrome).
Clinical features
The eye symptoms, termed keratoconjunctivitis sicca,
are due to a lack of lubricating tears, which, in turn,
reflects inflammatory infiltration of the lacrimal glands.
Conjunctivitis and blepharitis are frequent, and may
lead to filamentary keratitis due to tenacious mucous
filaments binding to the cornea and conjunctiva. Oral
involvement manifests as a dry mouth and typically the
patient needs to sip water to swallow food. There is a
high incidence of dental caries.
Other sites of extraglandular involvement are listed in Box. The disease is associated with a 40-fold increased lifetime risk of lymphoma.
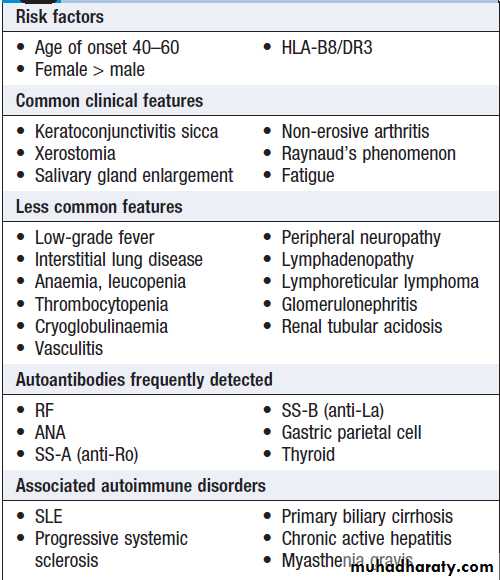
Features of Sjِgren’s syndrome
InvestigationsThe diagnosis can be established by the Schirmer tear
test, which measures tear flow over 5 minutes using
absorbent paper strips placed on the lower eyelid; a normal result is more than 6 mm of wetting. Staining
with rose Bengal may show punctate epithelial abnormalities over the area not covered by the open eyelid. If the diagnosis remains in doubt, it can be confirmed by lip biopsy, which shows focal lymphocytic infiltrate of the minor salivary glands. Most patients have an elevated ESR and hypergammaglobulinaemia, and one or more autoantibodies, including ANA and RF. Anti-Ro
and anti-La antibodies are commonly present.
Management
No treatments that have disease-modifying effects have
yet been identified and management is symptomatic.
Lachrymal substitutes, such as hypromellose, should be
used during the day in combination with more viscous
lubricating ointment at night. Soft contact lenses can be
useful for corneal protection in patients with filamentary
keratitis, and occlusion of the lachrymal ducts is occasionally needed. Artificial saliva and oral gels can be tried for xerostomia but are often not effective. Stimulation of saliva flow by sugar-free chewing gum or lozenges may be helpful.
Adequate post-prandial oral hygiene and prompt treatment of oral candidiasis are essential.
Vaginal dryness is treated with lubricants such
as K-Y jelly. Extraglandular and musculoskeletal manifestations may respond to steroids, and if so, immunosuppressive drugs can be added for their steroid-sparing effect.
Fatigue is difficult to treat; this is usually due to
non-restorative sleep (often because of xerostomia) and
is unresponsive to steroids. Immunosuppression does
not improve sicca symptoms. If lymphadenopathy or
salivary gland enlargement develops, biopsy should be
performed to exclude malignancy.
Polymyositis and dermatomyositis
Polymyositis and dermatomyositis are related disordersthat are characterised by an inflammatory process affecting skeletal muscle.
In dermatomyositis, characteristic skin changes also occur. Both diseases are rare, with an incidence of 2–10 cases per million/year. They can occur in isolation or in association with other autoimmune diseases, such as SLE, systemic sclerosis and Sjِgren’s syndrome.
The cause is unknown, although there is evidence for a genetic contribution.
Clinical features
The typical presentation of polymyositis is with
symmetrical proximal muscle weakness, usually affecting
the lower limbs more than the upper. The onset is usually between 40 and 60 years of age and is typically gradual, over a few weeks. Myositis is usually widespread but focal disease can also occur. Affected patients report difficulty rising from a chair, climbing stairs and lifting, sometimes in combination with muscle pain. Systemic features of fever, weight loss and fatigue are common. Respiratory or pharyngeal muscle involvement can lead to ventilatory failure or aspiration that requires urgent treatment. Interstitial lung disease occurs in up to 30% of patients and is strongly associated with the presence of antisynthetase (Jo-1) antibodies.
Dermatomyositis presents similarly but in combination
with characteristic skin lesions. These includeGottron’s papules, which are scaly, erythematous or violaceous, psoriaform plaques occurring over the extensor surfaces of PIP and DIP joints, and a heliotrope rash that is a violaceous discoloration of the eyelid in combination with periorbital oedema . Similar rashes occur on the upper back, chest and shoulders (‘shawl’ distribution). Periungual nail-fold capillaries are often enlarged and tortuous. There is about a threefold increased risk of malignancy in patients with dermatomyositis and polymyositis. This may be apparent at the time of presentation, but the risk remains increased for at least 5 years following diagnosis.
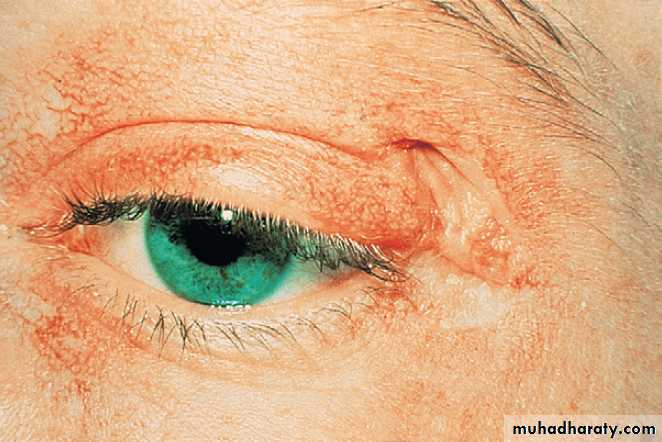
Typical eyelid appearance in dermatomyositis. Note the oedema and telangiectasia.
InvestigationsMuscle biopsy is the pivotal investigation and shows
the typical features of fibre necrosis, regeneration and
inflammatory cell infiltrate. Occasionally, however, a biopsy may be normal, particularly if myositis is patchy. In such cases, MRI is used to identify areas of abnormal muscle for biopsy. Serum levels of CK are usually raised and are a useful measure of disease activity,
although a normal CK does not exclude the diagnosis,
particularly in juvenile myositis. EMG can confirm
the presence of myopathy and exclude neuropathy. Screening for underlying malignancy should be undertaken routinely, and should include CT of chest/ abdomen/pelvis, upper and lower gastrointestinal endoscopy, and mammography in women.
Management
Oral corticosteroids (prednisolone 1 mg/kg daily) are
the mainstay of initial treatment, but high-dose intravenous
methylprednisolone (1 g/day for 3 days) may be
required in patients with respiratory or pharyngeal
weakness. If there is a good response, steroids should be
dose of 5–7.5 mg. Although most patients respond
well to corticosteroids, many need additional immunosuppressive therapy.
Azathioprine and methotrexate are the agents of first choice, but ciclosporin, cyclophosphamide, tacrolimus or MMF can be used as alternatives.
IV immunoglobulin may be effective in refractory cases. If the patient relapses or fails to respond to treatment,
this may be due to steroid-induced myopathy. A further
biopsy should be performed and may show type 2 fibre
atrophy in steroid-induced myopathy, as opposed to
necrosis and regeneration in active myositis.
VASCULITIS
These are a heterogeneous group of diseases characterised by inflammation and necrosis of blood-vessel walls, with associated damage to skin, kidney, lung, heart, brain and GIT . There is a wide spectrum of involvement and disease severity, ranging from mild and transient disease affecting only the skin, to life-threatening fulminant disease with multiple organ failure.
The clinical features result from a combination of local tissue ischaemia (due to vessel inflammation and narrowing) and the systemic effects of widespread inflammation.
Systemic vasculitis should be considered in any patient with fever, weight loss, fatigue, evidence of multisystem involvement, rashes, raised inflammatory markers and abnormal urinalysis .
Takayasu’s disease
Takayasu’s disease predominantly affects the aorta, its
major branches and occasionally the pulmonary arteries.
The typical age at onset is between 25 and 30 years, with
an 8 : 1 female preponderance. It has a worldwide
distribution but is most common in Asia. Takayasu’s is
characterised by granulomatous inflammation of the
vessel wall, leading to vessel occlusion or weakening
of the vessel wall. It presents with claudication, fever, arthralgia and weight loss. The vessels most commonly
affected are the aorta and carotid, ulnar, brachial,
radial and axillary arteries.
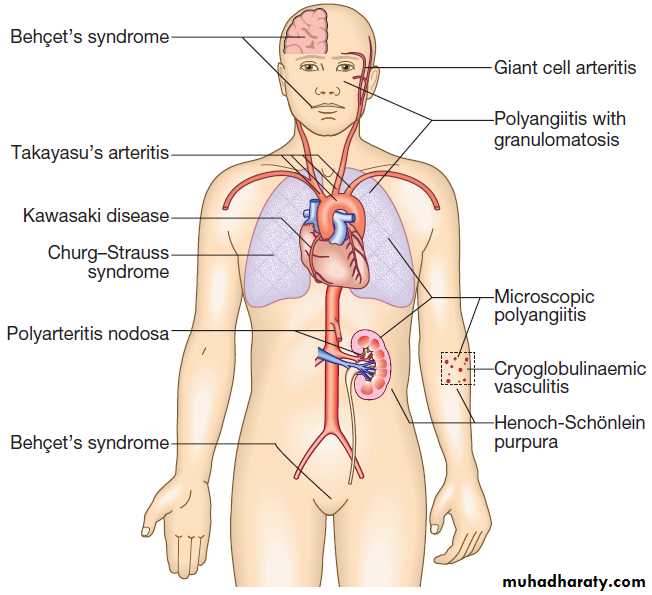
Types of vasculitis. The anatomical targets of different forms of vasculitis are shown.
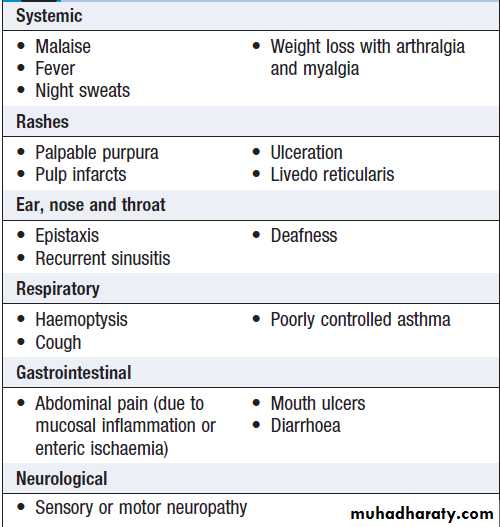
Clinical features of systemic vasculitis
Clinical examination may reveal loss of pulses, bruits, hypertension and aortic incompetence. Investigation will identify an acute phase response and normocytic, normochromic anaemia, but the diagnosis is based on angiography, which reveals coarctation, occlusion and aneurysmal dilatation. The distribution of involvement is classified into four types:
• type 1: localised to the aorta and its branches
• type 2: localised to the descending thoracic and
abdominal aorta
• type 3: combines features of 1 and 2
• type 4: involves the pulmonary artery.
Treatment
is with high-dose steroids and immunosuppressants, as described for SLE. With appropriate treatment, the 5-year survival is 83%.
Kawasaki disease
A vasculitis that mostly affects the coronary vessels. It presents as an acute systemic disorder, usually affecting children under 5 years. It occurs mainly in Japan. Presentation is with fever, generalised rash, including palms and soles, inflamed oral mucosa and conjunctival injection resembling a viral exanthem. The cause is unknown but is thought to be the result of an abnormal immune response to an infectious trigger.Cardiovascular complications include coronary arteritis, leading to MI , transient coronary dilatation, myocarditis, pericarditis, peripheral vascular insufficiency and gangrene. Treatment is with aspirin (5 mg/kg daily for 14 days) and IV gammaglobulin (400 mg/kg daily for 4 days).
Polyarteritis nodosa
Polyarteritis nodosa has a peak incidence between theages of 40 and 50, with a male preponderance of 2 : 1. Hepatitis B is an important risk factor, and the incidence is 10 times higher in the Inuit of Alaska, in whom hepatitis B infection is endemic. Presentation is with fever, myalgia,
arthralgia and weight loss, in combination with manifestations of multisystem disease. The most common
skin lesions are palpable purpura , ulceration, infarction and livedo reticularis . Pathological changes comprise necrotising inflammation and vessel occlusion, and in 70% of patients, arteritis of the vasa nervorum leads to neuropathy, which is typically symmetrical and affects both sensory and motor function.
Severe hypertension and/or renal impairment may occur due to multiple renal infarctions, but glomerulonephritis is rare (in contrast to microscopic polyangiitis).
The diagnosis is confirmed by angiography,
which shows multiple aneurysms and smooth
narrowing of mesenteric, hepatic or renal systems, or by
muscle or sural nerve biopsy, which reveals the histological
changes above. Treatment is with high-dose steroids
and immunosuppressants, as described for SLE.
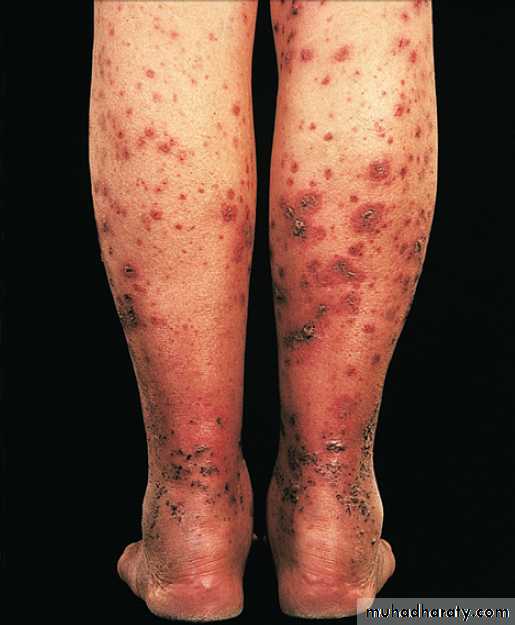
Rash of systemic vasculitis (palpable purpura).
Giant cell arteritis and polymyalgia rheumaticaGiant cell arteritis (GCA) is a granulomatous arteritis
that predominantly affects medium-sized arteries in the
head and neck. It is commonly associated with polymyalgia rheumatica (PMR), which presents with symmetrical muscle pain and stiffness affecting the shoulder and pelvic girdles. Since many patients with GCA have symptoms of PMR, and many patients with PMR go on to develop GCA if untreated, many rheumatologists
consider them to be different manifestations of the same underlying disorder. Both diseases are rare under the
age of 60 years. The average age at onset is 70, with a
female preponderance of about 3 : 1.
Clinical features
The cardinal symptom of GCA is headache, which isoften localised to the temporal or occipital region and
may be accompanied by scalp tenderness. Jaw pain
develops in some patients, brought on by chewing or
talking, due to ischaemia of the masseter muscles. Visual
disturbance can occur and a catastrophic presentation is
with blindness in one eye due to occlusion of the posterior ciliary artery. On fundoscopy, the optic disc may
appear pale and swollen with haemorrhages, but these
changes may take 24–36 hours to develop and the fundi
may initially appear normal. Other visual symptoms
include loss of visual acuity, reduced colour perception
and papillary defects.
Rarely, neurological involvement may occur, with transient ischaemic attacks, brainstem infarcts and hemiparesis.
The cardinal features of PMR are symmetrical muscle
pain and stiffness affecting the shoulder and pelvic
girdles. Constitutional symptoms, such as weight loss,
fatigue, malaise and night sweats, are common. The
onset of symptoms is usually fairly sudden over a few
days, but may be more insidious.
On examination, there may be stiffness and painful restriction of active shoulder movement but passive movements are preserved.
Muscles may be tender to palpation but weakness and
muscle-wasting are absent.
Conditions that can mimic polymyalgia rheumatic
• Fibromyalgia• Hypothyroidism
• Cervical spondylosis
• RA
• Systemic vasculitis
• Inflammatory myopathy
(particularly inclusion body myositis)
• Malignancy
Investigations
The typical laboratory abnormality is an elevated ESR,
often with a normochromic, normocytic anaemia. CRP
may also be elevated and in some cases this precedes
elevation of the ESR. Rarely, PMR and GCA can present
with a normal ESR. The diagnosis is usually based on a
combination of the typical clinical features, raised ESR
and prompt response to steroid. However, if there is
doubt concerning the diagnosis of GCA, a temporal
artery biopsy may be undertaken. Characteristic biopsy
findings are fragmentation of the internal elastic lamina
with necrosis of the media in combination with a mixed
inflammatory cell infiltrate.
Whilst a positive biopsy is helpful, a negative biopsy does not exclude the diagnosis because the lesions are focal. Ultrasound or arteriography may be used to help guide the biopsy.
Management
Corticosteroids are the treatment of choice and should
be commenced urgently in suspected GCA because of
the risk of visual loss . Response to treatment is dramatic, such that symptoms will have completely resolved within 48–72 hours of starting corticosteroid therapy in virtually all patients. It is customary to use higher doses in GCA (60–80 mg prednisolone) than in PMR (15–30 mg), although the evidence base for this is weak.
In both conditions the steroid dose should be progressively reduced, guided by symptoms and ESR, with the aim of reaching a dose of 10–15 mg by about 8 weeks. Thereafter, the rate of reduction should be slower – by 1 mg per month – until an acceptable dose is achieved (5–7.5 mg daily). If symptoms recur, the dose should be increased to that which previously controlled the symptoms, and reduction attempted again in another few weeks. Most patients need steroids for an average of 12–24 months. Some patients also require steroidsparing
agents, such as methotrexate or azathioprine, if
they require a maintenance dose of prednisolone of
more than 7.5 mg daily. Prophylaxis against osteoporosis
should be given in patients with low BMD.

Emergency management of giant cell arteritis
Antineutrophil cytoplasmic antibody-associated vasculitis (ANCA)Is a life-threatening disorder characterised by inflammatory infiltration of small blood vessels,
fibrinoid necrosis and the presence of circulating antibodies to ANCA. Two subtypes are recognised. Microscopic polyangiitis (MPA) is a necrotising small-vessel vasculitis found with rapidly progressive glomerulonephritis, often in association with alveolar haemorrhage. Cutaneous and gastrointestinal involvement is common and other features include neuropathy (15%) and pleural effusions (15%). Patients are usually myeloperoxidase (MPO) antibody-positive.
In granulomatosis with polyangiitis (also known as Wegener’s granulomatosis (WG)), the vasculitis is characterised by granuloma formation, mainly affecting the nasal passages, airways and kidney. A minority of patients present with glomerulonephritis. The most common presentation of WG is with epistaxis, nasal crusting and sinusitis, but haemoptysis and mucosal ulceration may also occur. Deafness may be a feature due to inner ear involvement, and proptosis may occur because of inflammation of the retro-orbital tissue .
This causes diplopia due to entrapment of the extra-ocular muscles, or loss of vision due to optic nerve compression. Disturbance of colour vision is an early feature of optic nerve compression.
Untreated nasal disease ultimately leads to destruction of bone and cartilage.
Migratory pulmonary infiltrates and nodulesoccur in 50% of patients. Patients with WG are usually
proteinase-3 (PR3) antibody-positive.
Patients with active disease usually have a leucocytosis
with an elevated CRP and ESR, in association
with raised ANCA levels. Complement levels are usually
normal or slightly elevated. Imaging of the upper
airways or chest with MRI can be useful in localising
abnormalities but, where possible, the diagnosis should
be confirmed by biopsy of the kidney or lesions in the
sinuses and upper airways.
Management is with high-dose steroids and
cyclophosphamide, as described for SLE, followed
by maintenance therapy with lower-dose steroids and
azathioprine, methotrexate or mycophenolate mofetil
(MMF). Rituximab in combination with high-dose steroids
is equally effective as oral cyclophosphamide at
inducing remission in ANCA-associated vasculitis. Both
MPA and WG have a tendency to relapse, and patients
must be followed on a regular and long-term basis to
check for clinical signs of recurrence. Measurements of
ESR, CRP and levels of ANCA antibodies are useful in
monitoring disease activity.
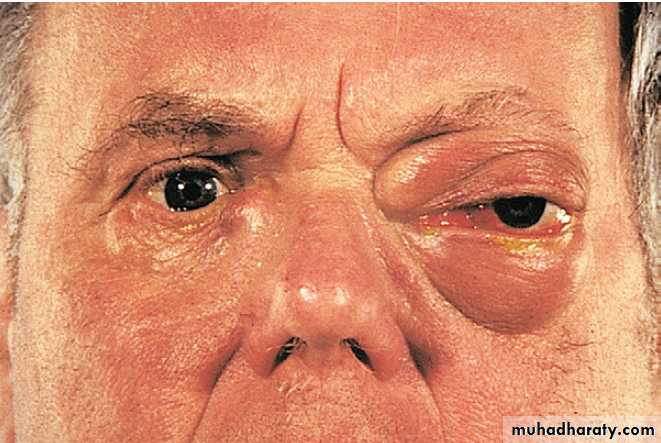
Eye involvement in granulomatosis with polyangiitis.
Churg–Strauss syndromeChurg–Strauss syndrome (CSS) is a small-vessel vasculitis
with an incidence of about 1–3 per 1 000 000; it is
associated with eosinophilia. Some patients have a prodromal period for many years, characterised by allergic rhinitis, nasal polyposis and late-onset asthma that is often difficult to control. The typical acute presentation is with a triad of skin lesions (purpura or nodules), asymmetric mononeuritis multiplex and eosinophilia.
Pulmonary infiltrates and pleural or pericardial effusions
due to serositis may be present. Up to 50% of
patients have abdominal symptoms provoked by
mesenteric vasculitis.
Patients with active disease have raised levels of ESR and CRP and an eosinophilia.
Although antibodies to MPO or PR3 can be detected in
up to 60% of cases, CSS is considered to be a distinct
disorder from the other ANCA-associated vasculitides.
Biopsy of an affected site reveals a small-vessel vasculitis
with eosinophilic infiltration of the vessel wall.
Management is with high-dose steroids and cyclophosphamide, followed by maintenance therapy with lowdose steroids and azathioprine, methotrexate or MMF.
Henoch–Schِnlein purpura (HSP)
Is a small-vessel vasculitis caused by immune complex deposition following an infectious trigger. It is predominantly a disease of children and young adults. The usual presentation is with purpura over the buttocks and lower legs, accompanied by abdominal pain, gastrointestinal bleeding and arthralgia. Nephritis can also occur and may present up to 4 weeks after the onset of other symptoms. Biopsy of affected tissue shows a vasculitis with IgA deposits in the vessel wall. HSP is usually a self-limiting disorder that settles spontaneously without specific treatment. Corticosteroids and immunosuppressive therapy may be required with more severe disease, particularly in the presence of nephritis.Cryoglobulinaemic vasculitis
This is a small-vessel vasculitis that can develop in some patients with circulating cryoglobulins, which are immunoglobulins that precipitate out in the cold. Cryoglobulins are classified into three types and types II and III are associated with vasculitis. The typical presentation is with a vasculitic rash over the lower limbs, arthralgia, Raynaud’s phenomenon and neuropathy. Some cases are secondary to hepatitis infection and others are secondary to other autoimmune diseases. Affected patients should be screened for evidence of hepatitis B and C infection, and if the results are positive, these should be treated appropriately .There is no consensus as to how best to treat cryoglobulinaemic vasculitis in the absence of an obvious trigger. Corticosteroids and immunosuppressive therapy are often used empirically but their efficacy is uncertain.
Behçet’s syndrome
This is a vasculitis of unknown aetiology that characteristically targets small arteries and venules. It is rare in Western Europe but more common in ‘Silk Route’ countries around the Mediterranean and Japan, where there is a strong association with HLA-B51.Oral ulcers are universal . Unlike aphthous
ulcers, they are usually deep and multiple, and last for
10–30 days. Genital ulcers are also a common problem,
occurring in 60–80% of cases. The usual skin lesions are
erythema nodosum or acneiform lesions, but migratory
thrombophlebitis and vasculitis also occur. Ocular
involvement is common and may include anterior or
posterior uveitis or retinal vasculitis.
Neurological involvement occurs in 5% and mainly involves the brainstem, although the meninges, hemispheres and
cord can also be affected, causing pyramidal signs,
cranial nerve lesions, brainstem symptoms or hemiparesis.
Recurrent thromboses also occur. Renal involvement
is extremely rare.
The diagnosis is primarily made on clinical grounds
(Box) but one characteristic feature that can be of
diagnostic value is the pathergy test, which involves
pricking the skin with a needle and looking for evidence
of pustule development within 48 hours.
Oral ulceration can be managed with topical steroid
preparations (soluble prednisolone mouthwashes, steroidpastes). Colchicine can be effective for erythema
nodosum and arthralgia. Thalidomide (100–300 mg per
day for 28 days initially) is very effective for resistant
oral and genital ulceration but is teratogenic and neurotoxic.
Steroids and immunosuppressants are indicated
for uveitis and neurological disease.

Criteria for the diagnosis of Behçet’s syndrome
Oral ulceration in Behçet’s syndrome
DISEASES OF BONEOsteoporosis
Osteoporosis is the most common bone disease and
affects millions of people worldwide. Fractures related
to osteoporosis are estimated to affect around 30% of
women and 12% of men in developed countries, and are
a major public health problem. In the UK alone, fractures
are sustained by over 250 000 individuals annually. Osteoporotic fractures can affect any bone, but the most common sites are the forearm (Colles fracture), spine (vertebral fracture) and hip .Of these, hip fractures are the most serious. Their immediate mortality is about 12% and there is a continued increase in mortality of about 20% when compared with age-matched controls.
Treatment of hip fracture accounts for the majority of the healthcare costs associated with osteoporosis.
The defining feature of osteoporosis is reduced bone
density, which causes a micro-architectural deterioration
of bone tissue and leads to an increased risk of
fracture. The prevalence of osteoporosis increases
with age, reflecting the fact that bone density declines
with age, especially in women .
The age-related decline in bone mass is accompanied by an increased risk of fractures .This is due in
part to the fall in bone density, but more importantly,
to the increased risk of falling, which increases with age.
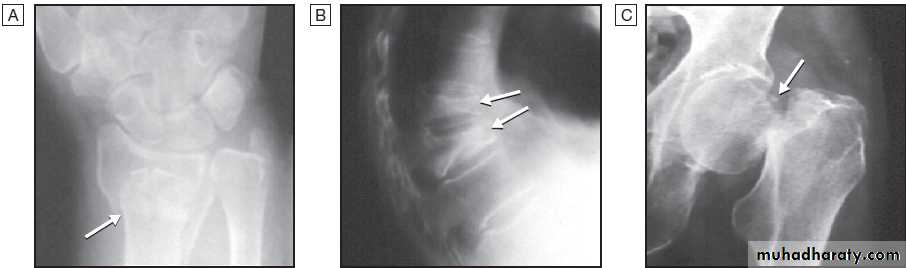
Osteoporotic fractures: X-rays.
A Wrist (Colles fracture).B Spine.
C Hip.
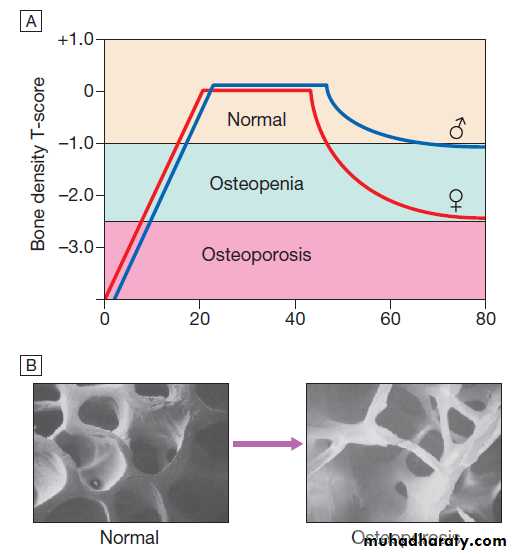
Changes in bone mass and microstructure with age.
A Changes in bone mass with age in men (blue line) and women (red line).B Scanning electron micrographs of normal bone (left) and
osteoporotic bone (right).
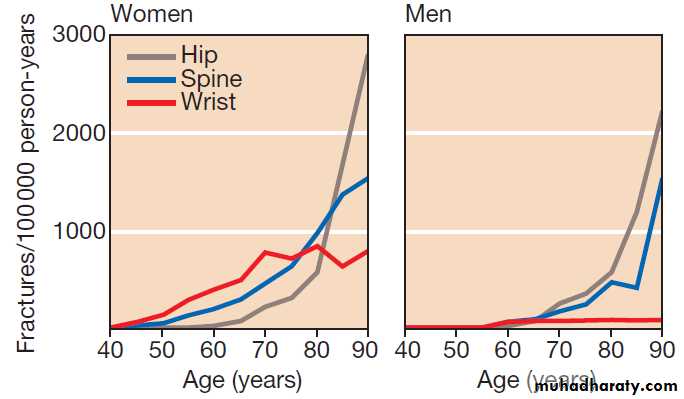
Relationship between age and the incidence of
osteoporotic fractures. Changes in the incidence of wrist fractures,vertebral fractures and hip fractures with age.
Pathophysiology
Osteoporosis occurs because of a defect in attaining peak
bone mass and/or because of accelerated bone loss. In
normal individuals, bone mass increases during skeletal
growth to reach a peak between the ages of 20 and
40 years but falls thereafter . In women there is an accelerated phase of bone loss after the menopause
due to oestrogen deficiency, which causes uncoupling
of bone resorption and bone formation, such that
the amount of bone removed by osteoclasts exceeds the
rate of new bone formation by osteoblasts. Age-related
bone loss is a distinct process that accounts for the
gradual bone loss that occurs with advancing age.
Bone resorption is not particularly increased but bone formation is reduced and fails to keep pace with bone resorption. Accumulation of fat in the bone marrow space also occurs because of an age-related decline in the ability of bone marrow stem cells to differentiate into osteoblasts and an increase in their ability to differentiate into adipocytes. Peak bone mass and bone loss are regulated by both genetic and environmental factors. Genetic factors account for up to 80% of the population variance in peak bone mass and other determinants of fracture risk, such as bone turnover and bone size. Polymorphisms have been identified in several genes that contribute to the pathogenesis of osteoporosis and many of these are in the RANK and Wnt signalling pathways, which play acritical role in regulating bone turnover.
Environmental factors, such as exercise and calcium
intake during growth and adolescence, are important inmaximising peak bone mass and in regulating rates of
post-menopausal bone loss.
Smoking has a detrimental effect on bone mineral density (BMD) and is associated with an increased fracture risk, partly because female smokers have an earlier menopause than non-smokers. Heavy alcohol intake is a recognised cause of osteoporosis and fractures.
Post-menopausal osteoporosis
This is the most common cause of osteoporosis because
of the effects of oestrogen deficiency, as described above. Early menopause (below the age of 45 years) is a particularly important risk factor.
Male osteoporosis
Osteoporosis is less common in men and a secondary
cause can be identified in about 50% of cases. The most common are hypogonadism, corticosteroid use (see below) and alcoholism. In hypogonadism, the pathogenesis is as described for post-menopausal osteoporosis, as testosterone deficiency results in an increase in bone turnover and uncoupling of bone resorption from bone formation. Genetic factors are probably important in the 50% of cases with no identifiable cause.
Corticosteroid-induced osteoporosis
This is an important cause of osteoporosis that relates todose and duration of therapy. Although there is no ‘safe’ dose of corticosteroid, the risk increases when the dose of prednisolone exceeds 7.5 mg daily and is continued for more than 3 months. Corticosteroids have adverse effects on calcium metabolism and bone cell function. A key abnormality is reduced bone formation due to a direct inhibitory effect on osteoblast function and steroid-induced osteoblast and osteocyte apoptosis. Corticosteroids also inhibit intestinal calcium absorption and cause a renal leak of calcium, and this tends to reduce serum calcium, leading to secondary hyperparathyroidism with increased osteoclastic bone resorption. Hypogonadism may also occur with high-dose steroids.
Pregnancy-associated osteoporosis
This is a rare presents with back pain and multiple vertebral fractures during the second or third trimester. The cause is unknown but may relate to an exaggeration of the bone loss that normally occurs during pregnancy, in pre-existing low bone mass.Primary hyperparathyroidism causes bone loss because sustained elevation in PTH increases bone turnover, and bone formation cannot keep pace with resorption. A similar mechanism operates in thyrotoxicosis, driven by raised levels of thyroid hormones.
Cushing’s disease is a rare cause, identical in mechanism to corticosteroid- induced osteoporosis.
Anorexia nervosa causes osteoporosis through calcium deficiency, low body weight and hypogonadism
Malabsorption predisposes to it through malnutrition, calcium and vitamin D deficiency and consequent secondary hyperparathyroidism.
Chronic HIV infection predisposes to osteoporosis because of low body weight, chronic immune activation and antiretroviral therapy. Inflammatory diseases increase bone resorption and suppress bone formation through release of pro-inflammatory cytokines such as IL-1 and TNF, and increased expression of RANK by lymphocytes. Similar mechanisms operate in certain cancers, which release a variety of bone-resorbing factors, including TNF, lymphotoxin and parathyroid
hormone-related protein (PTHrP).
Gaucher’s disease and systemic mastocytosis also cause release of bone resorbing factors.
Thiazolidinediones such as rosiglitazone inhibit osteoblast differentiation and promote adipocyte differentiation in the bone marrow, leading to reduced bone formation and bone loss.
Aromatase inhibitors cause osteoporosis by reducing peripheral conversion of adrenal androgens to oestrogen, whereas gonadotrophin-releasing hormone agonists do so by causing hypogonadism.
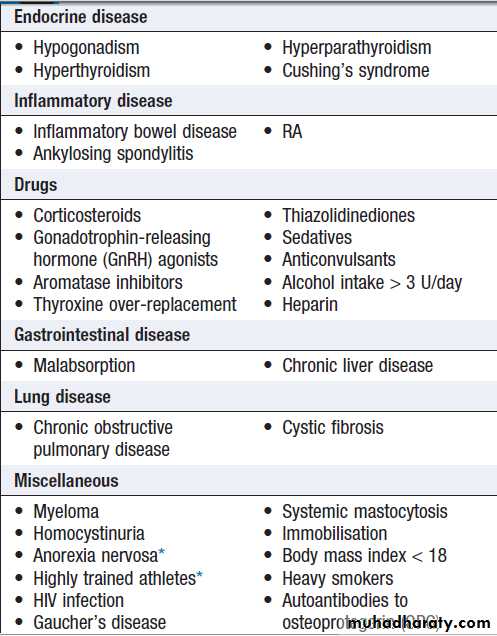
Secondary causes of osteoporosis and
osteoporotic fracturesClinical features
Patients with osteoporosis are asymptomatic until a fracture occurs. Osteoporotic spinal fracture may presentwith acute back pain or gradual onset of height loss and
kyphosis with chronic pain. The pain of acute vertebral
fracture can occasionally radiate to the anterior chest or
abdominal wall and be mistaken for a myocardial infarction or intra-abdominal pathology, but worsening of
pain by movement and local tenderness both suggest
vertebral fracture. Peripheral osteoporotic fractures
present with local pain, tenderness and deformity, often
after an episode of minimal trauma. In patients with hip
fracture, the affected leg is shortened and externally
rotated. Many patients present with incidental osteopenia
on an X-ray performed for other reasons.
Investigations
The pivotal investigation is dual energy X-ray absorptiometry (DEXA) at the lumbar spine and hip . This should be considered in patients with clinical risk factors for osteoporosis , and those with an elevated 10-year fracture risk as defined by a fracture risk assessment tool. Figure . provides an algorithm for the investigation
of patients with suspected osteoporosis based on clinical
risk factors, fracture risk assessment and DEXA.
A history should be taken to identify any predisposing
causes, such as early menopause, excessive alcohol
intake, smoking and corticosteroid therapy. Signs of
endocrine disease, neoplasia and inflammatory disease
should be sought on clinical examination.
A falls history should be taken and a ‘get up and go’ test performed, especially in older patients . Renal function, liver function, thyroid function, immunoglobulins and ESR,
with screening for coeliac disease (anti-tissue transglutaminase (tTG) antibodies), should be performed.
Serum 25(OH) vitamin D and PTH measurements are
useful to exclude vitamin D deficiency and secondary
hyperparathyroidism. Primary hyperparathyroidism
should be suspected if hypercalcaemia is present.
Levels of sex hormones and gonadotrophins should be measured in men with osteoporosis and women under the age of 50. Transiliac bone biopsy is sometimes required in early-onset osteoporosis of unknown cause or when coexisting osteomalacia is suspected.

Indications for bone densitometry
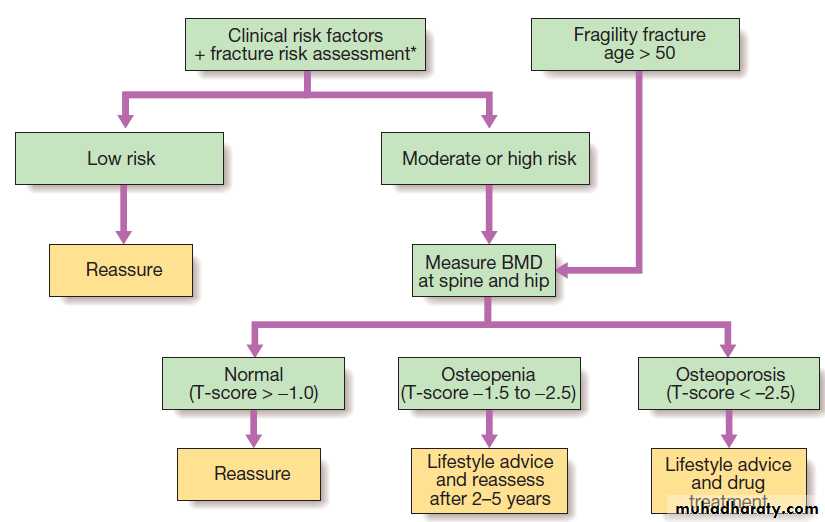
Algorithm for the investigation of patients with suspected osteoporosis. *Using FRAXR or QFracture,
(BMD = bone mineral density)
Management
The aim of treatment is to reduce the risk of fractureand this can be achieved by a combination of nonpharmacological and pharmacological approaches.
Non-pharmacological interventions
Advice on smoking cessation, moderation of alcohol
intake, adequate dietary calcium intake and exercise
should be given. Those with recurrent falls or unsteadiness
on a ‘get up and go’ test should be referred to a
multidisciplinary falls prevention team . Hip protectors can reduce the risk of hip fracture in selected patients but adherence is often poor.
Drug treatment
Several drugs have been shown to reduce the risk ofosteoporotic fractures in randomised controlled trials.
Their effects on vertebral and non-vertebral fracture are
also summarised in Box.
Drug treatment should be considered in patients
with BMD T-score values below −2.5 or below −1.5 in
corticosteroid-induced osteoporosis, because there is
evidence that fractures occur at a higher BMD value in
steroid users and that drugs prevent fracture in patients
with T-scores at this level. Treatment should also be
considered in patients with vertebral fractures, irrespective
of BMD, unless they resulted from significant trauma.
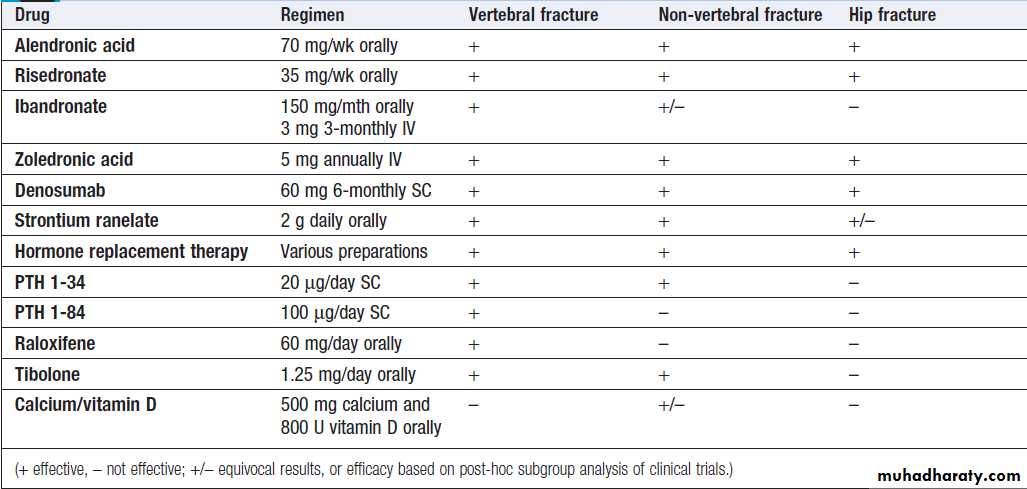
Effects of drugs on the risk of osteoporotic fractures
BisphosphonatesBisphosphonates inhibit bone resorption by binding to
hydroxyapatite crystals on the bone surface. When
Osteoclasts attempt to resorb bone that contains bisphosphonate, the drug is released within the cell, where it inhibits key signalling pathways that are essential for
osteoclast function. Although bisphosphonates primarily
target the osteoclast, bone formation is also suppressed
because of coupling between bone formation
and bone resorption and an inhibitory effect on osteoblasts.
However, the balance of effect on bone turnover
is favourable, resulting in a gain in bone density due
partly to increased mineralisation of bone.
Bisphosphonate treatment typically leads to an increase in spine BMD of about 5–8% and in hip BMD of 2–4% during the first 3 years of treatment and plateaus thereafter.
Alendronic acid is the bisphosphonate used most frequently.
It reduces risk of vertebral fractures by 40% and
non-vertebral fractures by about 25% in postmenopausal
women with osteoporosis. Risedronate is an alternative
with similar efficacy, but may be better tolerated in
patients with a history of gastrointestinal upset. Both
drugs are effective in the treatment of corticosteroid-induced osteoporosis.
They can also be used to treat male osteoporosis but neither has been shown to prevent non-vertebral fractures in men. Ibandronate is sometimes used but the evidence for prevention of nonvertebral fractures is less robust. Zoledronic acid is effective in the treatment of post-menopausal osteoporosis, corticosteroid-induced osteoporosis and osteoporosis in men.
It reduces the risk of vertebral fracture by about 75% with similar effects to alendronic acid on nonvertebral fractures.
It is especially useful for secondary prevention of fractures in elderly patients with hip fracture and reduces mortality in this group, being the only treatment that has been shown to modify this. Etidronate reduces the risk of vertebral fracture but the effects on non-vertebral fracture are less robust than those of other bisphosphonates. It is now seldom used.
Oral bisphosphonates are poorly absorbed from the
gastrointestinal tract and should be taken on an empty
stomach with plain water; no food should be eaten
for 30–45 minutes after administration.
Upper gastrointestinal upset occurs in about 5% so oral bisphosphonates should be used with caution in patients with existing gastro-oesophageal reflux disease. They should be avoided in patients with oesophageal stricture or achalasia, since tablets may stick in the oesophagus,
causing ulceration and perforation.
The most common adverse effect with intravenous bisphosphonates is a transient influenza-like illness characterised by fever, malaise, anorexia and generalised aches, which occurs 24–48 hours after administration.
This is self-limiting but can be treated with paracetamol or NSAID if necessary.
It predominantly occurs after the first exposure and tolerance develops thereafter. Other adverse effects areshown in Box. Osteonecrosis of the jaw (ONJ) is
characterised by the presence of necrotic bone in the
mandible or maxilla, typically occurring after tooth
extraction when the socket fails to heal. Most ONJ cases
have occurred in cancer patients with coexisting morbidity,
such as infection and diabetes, who have received
high doses of intravenous bisphosphonates; this complication is very rare in patients who are treated with the dose regimes used in osteoporosis.
None the less, all patients receiving bisphosphonates for any reason should be advised to pay attention to good oral hygiene.
There is no evidence that temporarily stopping medication
in patients undergoing tooth extraction is necessary
or alters the occurrence of ONJ. Atypical subtrochanteric
fractures have been described in patients who have
received long-term bisphosphonates, and may be the
result of over-suppression of normal bone remodelling.
In the vast majority, the benefits of bisphosphonate
therapy far outweigh the risks, but it is important that
treatment is targeted to patients with low BMD who are
most likely to benefit.

Adverse effects of bisphosphonates
DenosumabDenosumab is a monoclonal antibody that neutralises
the effects of RANKL , it is administered by subcutaneous injection every 6 months in the treatment of osteoporosis. It is a powerful inhibitor of bone resorption and reduces the risk of hip fractures by 40%, vertebral fractures by 70% and other non-vertebral fractures by 20%. It has few adverse effects but there are isolated reports of ONJ with long-term use.
Unlike bisphosphonates, its duration of action is short
and it must be administered on a long-term basis to
maintain its effect on bone mass and bone turnover.
Calcium and vitamin D
Calcium and vitamin D have limited efficacy in the
prevention of osteoporotic fractures when given in
isolation but are widely used as an adjunct to other
treatments, most often as combination preparations containing 500 mg calcium and 800 U vitamin D. They are
of greatest value in preventing fragility fractures in
elderly or institutionalised patients who are at high risk
of calcium and vitamin D deficiency .
Strontium ranelate
Strontium ranelate reduces vertebral fracture risk byabout 50% after 3 years and non-vertebral fracture risk
by 12%. The mechanism of action is poorly understood.
It has a weak inhibitory effect on bone resorption, stimulates biochemical markers of bone formation and is
incorporated within hydroxyapatite crystals in place of
calcium. Large changes in BMD (12%) occur, although
this is partly an artefact due to substitution of strontium
for calcium in bone mineral. The most common adverse
effect is diarrhoea. Strontium is contraindicated in
patients with cardiovascular disease due to an increased risk of myocardial infarction. There is also an increased
risk of venous thrombosis. Rarely, a severe rash occurs,
and this is an indication to stop treatment.
Parathyroid hormone
PTH is an anabolic agent that works by stimulating new
bone formation. The most widely used preparation is the
1-34 fragment of PTH (teriparatide) given by single daily
subcutaneous injection of 20 μg. Teriparatide increases
BMD by 10% or more in osteoporotic subjects and reduces risk of vertebral fractures by about 65% and non-vertebral fractures by 50%. It is also effective in corticosteroid-induced osteoporosis and appears superior to alendronate in terms of BMD gain and vertebral fracture reduction. It is also effective in male osteoporosis. PTH is expensive and is usually reserved for patients with severe osteoporosis (BMD T-score of −3.5 to −4.0 or below) and those who have failed to respond adequately to other treatments.
The recommended duration of treatment is 24 months, after which patients should receive an antiresorptive drug, such as a bisphosphonate, to maintain the increase in BMD. Teriparatide should not be administered at the same time as bisphosphonates, as this blunts the anabolic effect. In patients who are being treated with teriparatide because of failure to respond, existing treatments should be stopped. The 1-84 fragment of PTH acts in a similar way to teriparatide but the evidence for prevention of non-vertebral fracture is less robust.
Hormone replacement therapy, raloxifene and tibolone
Cyclical HRT with oestrogen and progestogen preventspost-menopausal bone loss and reduces the risk of vertebral and non-vertebral fractures in post-menopausal
women. It is primarily indicated for the prevention of
osteoporosis in women with an early menopause and for treatment of women with osteoporosis in their early fifties who have troublesome menopausal symptoms. HRT should be avoided in older women with established osteoporosis because it significantly increases the risk of breast cancer and cardiovascular disease. Raloxifene acts as a partial agonist at oestrogen receptors in bone and liver but as an antagonist in breast and endometrium, and is classified as a selective oestrogen receptor modulator (SERM).
It results in a modest increase in BMD (2%) and a 40% reduction in vertebral fractures, but does not influence the risk of non-vertebral fracture and can provoke muscle cramps and worsen hot flushes. It increases the risk of VTE to a similar extent as HRT but reduces the risk of breast
cancer; it does not influence the risk of cardiovascular
disease. Bazedoxifene is a related SERM that has similar
effects to raloxifene. Tibolone is a steroid that has partial
agonist activity at oestrogen, progestogen and androgen
receptors. It has similar effects on BMD to raloxifene and
has been found to prevent vertebral and non-vertebral
fractures in post-menopausal osteoporosis. Treatment is
associated with a slightly increased risk of stroke but a
reduced risk of breast cancer.
Other drugs
Calcitonin is an osteoclast inhibitor that has weak antifracture efficacy but is no longer used in the treatment of osteoporosis because of concerns about an increased risk of cancer with long-term use. It is occasionally used (unlicensed) in the short-term treatment of patients with acute vertebral fracture, when it is given by subcutaneous or intramuscular injection (100–200 U daily). Calcitriol (1,25(OH)2D3), the active metabolite of vitamin D, is licensed for treatment of osteoporosis, but it is seldom used since the data on fracture prevention are less robust than for other agents.
Duration of therapy and monitoring response
Oral bisphosphonates are usually given on a long-termbasis for osteoporosis with periodic review of the
continued need for therapy at 5-yearly intervals. The
evidence base on duration of treatment is limited. Alendronate and risedronate appear to be safe and effective for up to 10 years in most patients, although one randomized trial with alendronate showed that overall fracture rates were similar in those given 5 years’ therapy as opposed to 10. Studies with intravenous zoledronic acid
have shown that 3 years’ treatment give equal protection
from fractures as 6 years’ treatment. Other drug treatments, such as HRT, raloxifene and denosumab, need to be given continuously for a beneficial effect.
The optimal duration of treatment for strontium has not been established.
For anabolic drugs such as PTH, a 2-year courseof treatment is given and followed by long-term antiresorptive therapy.
The response to drug treatment can be assessed by
repeating BMD measurements after 2–3 years. However,
changes in BMD do not predict anti-fracture efficacy
well and there is little evidence that monitoring by BMD
or markers improves adherence.
It may, however, reassure the patient that the treatment is working. Since the precision of spine BMD (approximately 1%) is better than hip (approximately 2.5%), spine BMD is best for monitoring.
To be sure that a change has occurred, about twice the precision (about 2% for spine, 5% for hip) is required. This means that, under normal circumstances, at least 2 years should have elapsed before a repeat scan is performed. Biochemical markers of bone turnover,
such as N-telopeptide (NTX), respond more quickly
than BMD and can be used to assess adherence, but the
correlation with anti-fracture efficacy is modest. Treatment response can also be established by measuring
change in patient height (to assess progression of
vertebral osteoporosis) and documenting the occurrence
of clinical fractures.
Surgery
Orthopaedic surgery is frequently required to reduceand stabilise osteoporotic fractures. Patients with intracapsular fracture of the femoral neck generally require hemi-arthroplasty or total hip replacement in view of the high risk of avascular necrosis.
Vertebroplasty and kyphoplasty
Vertebroplasty (VP) is sometimes used in the treatment
of painful vertebral compression fractures. It involves
injecting methyl methacrylate (MMA) into the affected
vertebral body. Although VP has been found to give
better pain relief than medical therapy in the short term,
recent randomised controlled trials that compared VP
with a sham procedure showed no benefit , indicating that the reduction in pain may be a placebo response.
Kyphoplasty (KP) is used under similar circumstances
but in this case a needle is introduced into the affected vertebral body and a balloon is inflated, which is then filled with MMA. This procedure is more effective than medical treatment at relieving pain in the short term, with results similar to VP.The effects of KP have not so far been compared with a sham procedure.
Both procedures are generally safe but serious adverse
effects include spinal cord compression, and fat embolus
may occur.

Vertebroplasty in painful vertebral fractures
Management of recurrent fractureSince the treatments for osteoporosis are incompletely effective at preventing fracture, it is not uncommon to encounter patients who suffer recurrent fractures whilst on treatment. In these circumstances it is useful to perform DEXA to determine whether BMD has
increased, provided that sufficient time has elapsed to assess this (see above). If BMD has increased, then treatment should be continued. If there has been no BMD response or significant bone loss has occurred, the patient should be questioned about adherence and asked if the treatment is being taken correctly. This
is particularly important with oral bisphosphonates, where absorption is poor and is inhibited by food. If the patient appears to be taking the medication correctly and significant bone loss has occurred, then a different treatment should be considered.
Osteoporosis in old age
• Bone loss: due to increased bone turnover, with anage-related defect switch in differentiation of bone marrow
stromal cells to form adipocytes as opposed to osteoblasts.
• Fractures due to osteoporosis: common cause of morbidity
and mortality, although fracture healing is not delayed by age.
• Recurrent fractures: those who suffer a fragility fracture are
at increased risk of further fracture, so should be
investigated for osteoporosis and treated if this is confirmed.
• Falls: risk factors for falls (such as visual and neuromuscular
impairments) are independent risk factors for hip fracture in
elderly women, so intervention to prevent falls is as
important as treatment of osteoporosis .
• Intravenous zoledronic acid: reduces mortality and
subsequent fracture in elderly patients with hip fractures.
• Calcium and vitamin D: reduce the risk of fractures in those
who are housebound or living in care homes.
Osteomalacia and rickets
These conditions are characterised by defective mineralization of bone due to vitamin D deficiency, resistance to the effects of vitamin D or hypophosphataemia.
Osteomalacia describes a syndrome in adults of defective bone mineralisation, bone pain, increased bone fragility and fractures. Rickets is the equivalent syndrome
in children and is characterised by enlargement
of the growth plate and bone deformity. The disease
remains prevalent in frail older people who have a poor
diet and limited sunlight exposure, and in some Muslim
women who live in northern latitudes. There are four
main causes of osteomalacia and rickets (Box).
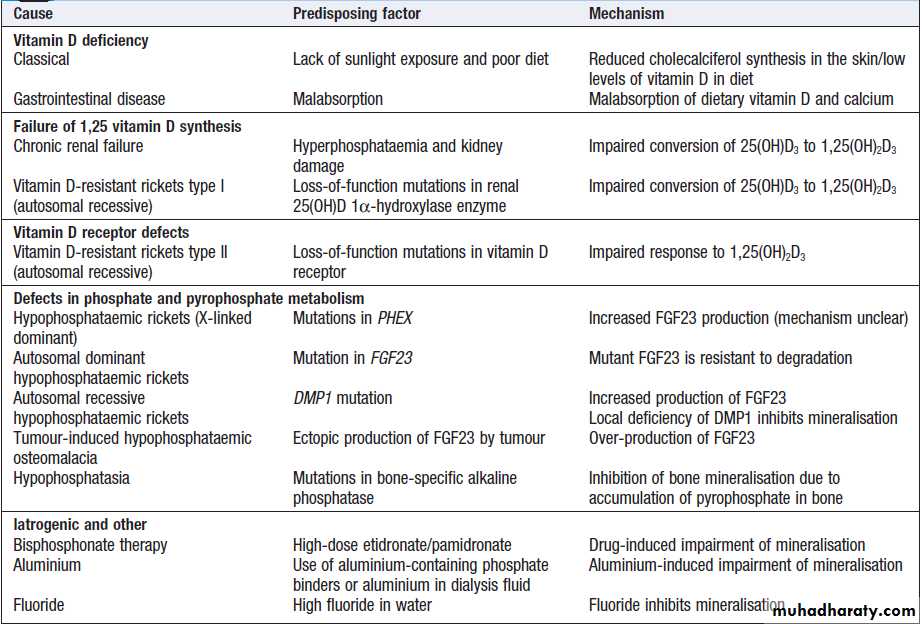
Causes of osteomalacia and rickets
Vitamin D deficiencyThe most common cause of osteomalacia and rickets is vitamin D deficiency, which can result from either lack of sunlight exposure, from which the majority of vitamin D is derived; dietary deficiency ; or malabsorption of vitamin D in patients with gastrointestinal disease.
Pathophysiology
The source of vitamin D and pathways involved inregulating its metabolism are shown in Figure. In
normal individuals, vitamin D (also known as cholecalciferol) comes from two sources: about 70% is made in the skin from 7-dehydrocholesterol under the influence of ultraviolet light, whereas the remaining 30% is derived from the diet. On entering the circulation,
vitamin D is hydroxylated in the liver to form 25(OH)
vitamin D and this is further hydroxylated in the kidney
to form 1,25(OH)2D, the biologically active metabolite.
The 1,25(OH)2D primarily acts on the gut to increase
intestinal calcium absorption but also acts on the
skeleton to stimulate bone remodelling.
Synthesis of 1,25(OH)2D is regulated by a negative feedback loop orchestrated by the parathyroid glands. When vitamin D levels fall – for example, as the result of reduced sunlight exposure or dietary lack – 25(OH)D and 1,25(OH)2D levels also fall, resulting in a reduction in calcium absorption from the gut. This causes serum calcium levels to fall, and this is detected by calcium-sensing receptors on the parathyroid chief cells, which respond by secreting parathyroid hormone (PTH). The raised levels of PTH restore calcium levels to normal by stimulating production of 1,25(OH)2D, reducing renal calcium excretion and increasing bone resorption.
Renal phosphate excretion also increases, lowering serum phosphate levels. Initially, these changes are effective in
maintaining normal levels of serum calcium but, with
prolonged vitamin D deficiency, reserves of 25(OH)D
become progressively depleted (through increased
conversion of 25(OH)D to 1,25(OH)2D), leading to
hypocalcaemia and hypocalcaemia with progressive
demineralisation of the skeleton and the clinical syndromes
of osteomalacia and rickets.
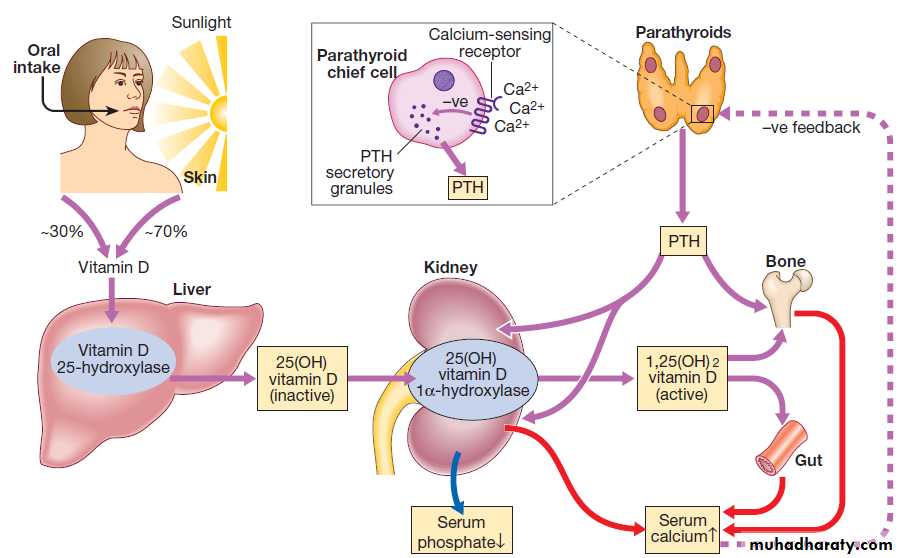
Vitamin D metabolism. There is close interaction between vitamin D, serum calcium and parathyroid hormone (PTH).
Clinical features
Vitamin D deficiency in children causes delayed development, muscle hypotonia, craniotabes (small unossified areas in membranous bones of the skull that yield to finger pressure with a cracking feeling), bossing of the frontal and parietal bones and delayed anterior fontanelle closure, enlargement of epiphyses at the lower end of the radius, and swelling of the rib costochondral junctions (‘rickety rosary’). Osteomalacia in adults presents insidiously. Mild osteomalacia can be asymptomatic or present with fractures and mimic osteoporosis.More severe osteomalacia presents with muscle and bone
pain, general malaise and fragility fractures. Proximalmuscle weakness is prominent and the patient may walk
with a waddling gait and struggle to climb stairs or get
out of a chair. There may be bone and muscle tenderness
on pressure and focal bone pain can occur due to fissure
fractures of the ribs and pelvis.
Investigations
The diagnosis can usually be made on a biochemical
screen with measurement of serum 25(OH)D and PTH.
Typically, serum ALP levels are raised, 25(OH)D levels
are low or undetectable, and PTH is elevated. Serum
calcium and phosphate levels may also be low but
normal values do not exclude the diagnosis. X-rays are
normal until advanced disease, when focal radiolucent
areas (pseudofractures or Looser’s zones) may be seen
in ribs, pelvis and long bones . Radiographic osteopenia is common and the presence of vertebral crush fractures may cause confusion with osteoporosis.
In children, there is thickening and widening of the
epiphyseal plate. Radionuclide bone scan can showmultiple hot spots in the ribs and pelvis at the site of
fractures and the appearance may be mistaken for
metastases.
Where there is doubt, the diagnosis can be confirmed by bone biopsy, which shows the pathognomonic features of increased thickness and extent of osteoid seams .
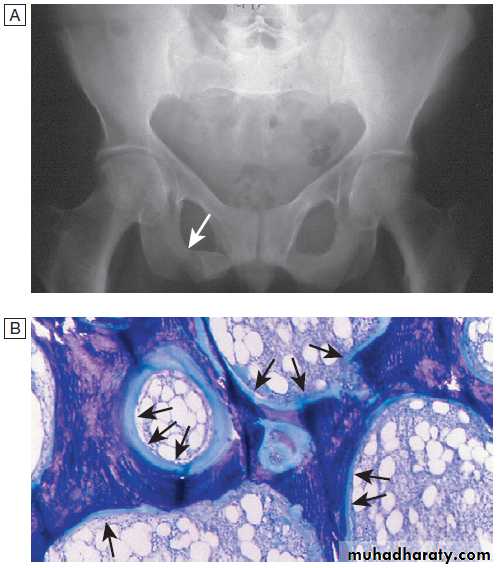
Osteomalacia.
A X-ray of the pelvis showing Looser’s zones (arrow).
B Photomicrograph of bone biopsy from osteomalacic patient showing thick osteoid seams (stained light blue) that cover almost
all of the bone surface. Calcified bone is stained dark blue.
Management
Osteomalacia and rickets respond promptly to treatmentwith vitamin D (250–1000 μg daily), with rapid clinical
improvement, an elevation in serum 25(OH)D and a
reduction in PTH. Serum ALP levels sometimes rise initially
as mineralisation of bone increases, but eventually fall to within the reference range as the bone disease
heals. After 3–4 months, treatment can generally be
stopped or the dose of vitamin D reduced to a maintenance level of 10–20 μg of cholecalciferol daily, except in patients with underlying disease such as malabsorption, in whom higher doses may be required.
Vitamin D-resistant rickets
The term vitamin D-resistant rickets (VDRR) describesosteomalacia and rickets caused by:
• inactivating mutations in the 25-hydroxyvitamin D
1α-hydroxylase (CYP27B1) enzyme, which converts
25(OH)D to the active metabolite 1,25(OH)2D3 (type I VDRR)
• inactivating mutations in the vitamin D receptor,
which impair its ability to activate transcription (type II VDRR). Clinical features are similar to those of infantile rickets and the diagnosis is usually first suspected when the patient fails to respond to vitamin D supplementation. Since both are recessive disorders, consanguinity is common but there may or may not be a positive family history.
Biochemical features of type I disease are similar to
vitamin D deficiency, except that levels of 25(OH)D are
normal.
In type II disease, 25(OH)D is normal but PTH
and 1,25(OH) 2D3 values are raised. Type I can be treated with the active vitamin D metabolites, 1α-hydroxyvitamin D (1–2 μg daily, orally) or 1,25 dihydroxyvitamin D (0.25–1.5 μg daily, orally), with or without calcium supplements, depending upon the patient’s diet.
Type II VDRR is extremely difficult to treat but sometimes
responds partially to very high doses of active vitamin D
metabolites and calcium and phosphate supplements.
Renal rickets and osteomalacia
Osteomalacia and rickets occur in patients with chronicrenal failure due to defects in synthesis of renal
1,25(OH) 2D3 or due to over-aggressive treatment with
oral phosphate binders.
Hypophosphataemic rickets and osteomalacia
Rickets and osteomalacia can occur as the result of
Inherited or acquired defects in renal tubular phosphate
reabsorption, and rarely in patients with tumours that
secrete phosphaturic substances .
Pathophysiology
Circulating levels of FGF23 play a critical role in regulating serum phosphate by modulating expression ofsodium-dependent phosphate transporters in the
kidney, which are responsible for renal tubular phosphate
reabsorption. Osteocytes are the main source of
FGF23 and levels of expression are regulated by the
proteins DMP1 and PHEX, which are also produced by
osteocytes . Inherited mutations affecting these proteins are summarised in Box and account for most cases of hypophosphataemic rickets.
Acquired hypophosphataemic rickets is mostly
caused by over-production of FGF23 by tumours.
Clinical features and diagnosis
The hereditary disorders usually present in childhood
with rickets. The diagnosis is made on the basis of the
early age at onset and the presence of hypophosphataemia with renal phosphate wasting, in the absence of vitamin D deficiency.
Molecular diagnosis can define the causal mutation. Tumour-induced hypophosphataemic osteomalacia presents with severe, rapidly progressive symptoms in patients with no obvious predisposing factor for osteomalacia. Strenuous efforts should be
made to identify the underlying, usually occult tumour
with whole-body MRI or CT.
Management
Treatment is with phosphate supplements (1–4 g daily)and active metabolites of vitamin D (1α-hydroxyvitamin
D, 1–2 μg daily, or 1,25 dihydroxyvitamin D, 0.25–1.5 μg
daily) to promote intestinal calcium and phosphate
absorption. The aim is to ameliorate symptoms, restore
normal growth, maintain serum phosphate levels within
the reference range and normalise ALP levels. Levels
of calcium, phosphate, ALP and renal function should
be monitored. Tumour-induced osteomalacia can be
managed in the same way but ideally should be treated
with surgical excision of the tumour since this is curative.
Hypophosphatasia
Hypophosphatasia is an autosomal recessive disorder
caused by inactivating mutations in the TNALP gene
that impair ALP function, resulting in accumulation of
pyrophosphate and inhibition of bone mineralisation.
Chondrocalcinosis may also occur. The diagnosis
should be suspected in osteomalacic patients with low
or undetectable levels of serum ALP but normal levels
of calcium, phosphate, PTH and vitamin D metabolites.
Medical treatment with injections of recombinant ALP
has recently been introduced with promising results.
Other causes of osteomalacia
These are summarised in Box. Aluminium intoxication is now rare due to reduced use of aluminium-containing phosphate binders and removal of aluminium from the water supplies used in dialysis. If aluminium intoxication is suspected, the diagnosis can be confirmedby demonstration of aluminium at the calcification front in a bone biopsy. Osteomalacia due to bisphosphonates has mostly been described in patients with Paget’s disease receiving etidronate and high-dose pamidronate.
It is usually asymptomatic and healing occurs when treatment is stopped. Excessive fluoride intake causes osteomalacia due to direct inhibition of mineralization and is common in parts of the world where there is a high fluoride content in drinking water. The condition reverses when fluoride intake is reduced.
Paget’s disease of bone
Paget’s disease of bone (PDB) is a common conditioncharacterised by focal areas of increased and disorganized bone remodelling. It mostly affects the axial skeleton, and bones that are commonly involved include the pelvis, femur, tibia, lumbar spine, skull and scapula. It is seldom diagnosed before the age of 40, but gradually increases in incidence thereafter to affect up to 8% of the UK population by the age of 85. The disease is common in Caucasians from north-west and southern Europe, supporting the importance of genetic factors in the aetiology, but the incidence of PDB has fallen in some countries over the past 25 years, suggesting that environmental triggers also play a role.
Pathophysiology
The primary abnormality is increased osteoclastic bone
resorption, accompanied by marrow fibrosis, increased
vascularity of bone and increased osteoblast activity.
Bone in PDB is architecturally abnormal and has reduced
mechanical strength.
Osteoclasts in PDB are increased in number, are unusually large and contain characteristic nuclear inclusion bodies. Genetic factors are important and mutations in the SQSTM1 gene are a common cause of classical PDB.
The presence of nuclear inclusion bodies in osteoclasts has fuelled speculation that PDB might be caused by a slow virus infection with measles or distemper but the evidence is conflicting. Biomechanical factors may help determine the pattern of involvement, since PDB often starts at sites of muscle insertions into bone and, in some cases, localises to bones or limbs that have been subjected to repetitive trauma or overuse.
Involvement of subchondral bone can compromise the
joint and predispose to OA (‘Pagetic arthropathy’).
Clinical features
The classic presentation is with bone pain, deformity,deafness and pathological fractures, but many patients
are asymptomatic and diagnosed from an abnormal
X-ray or blood test performed for another reason. Clinical
signs include bone deformity and expansion,
increased warmth over affected bones, and pathological
fracture. Bone deformity is most evident in weight-bearing
bones such as the femur and tibia, but when
the skull is affected the patient may complain that hats
no longer fit due to cranial enlargement.
Neurological problems, such as deafness, cranial nerve defects, nerve root pain, spinal cord compression and spinal stenosis, are recognised complications due to enlargement of affected bones and encroachment upon the spinal cord and nerve foraminae. Surprisingly, deafness seldom results from compression of the auditory nerve, but is conductive due to osteosclerosis of the temporal bone.
The increased vascularity of Pagetic bone makes operative procedures difficult and, in extreme cases, can precipitate high-output cardiac failure in elderly patients
with limited cardiac reserve. Osteosarcoma is a rare but
serious complication that presents with subacute onset
of increasing pain and swelling of an affected site.
Investigations
The characteristic features are an elevated serum ALPand bone expansion on X-ray, with alternating areas of
radiolucency and osteosclerosis . ALP is normal in about 5% of cases, usually because of monostotic
involvement. Radionuclide bone scanning is useful
to define the presence and extent of disease .
If the bone scan is positive, X-rays should be taken of an
affected bone to confirm the diagnosis. Bone biopsy is
not usually required but may help to exclude osteosclerotic
metastases in cases of diagnostic uncertainty.
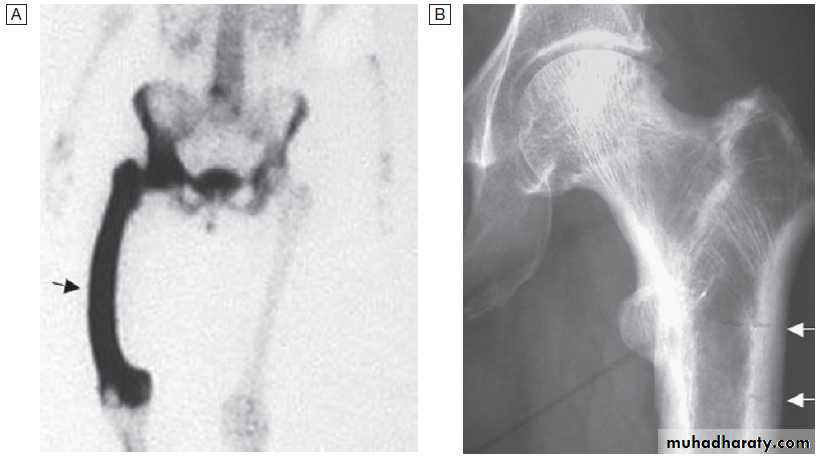
Paget’s disease.
A Isotope bone scan from a patient with Paget’s disease, illustrating the intense tracer uptake and deformity of the affected femur.B The typical radiographic features with expansion of the femur, alternating areas of osteosclerosis and radiolucency of the trochanter, and pseudofractures breaching the bone cortex (arrows).
Management
The main indication for treatment with inhibitors of bone resorption is bone pain thought to be due to increased metabolic activity .
It is often difficult to differentiatethis from pain due to complications such as bone deformity, nerve compression symptoms and OA.
If there is doubt, it can be worthwhile giving a therapeutictrial of antiresorptive therapy to determine whetherthe symptoms improve.
A positive response indicates that the pain was due to increased metabolic activity.
The aminobisphosphonates pamidronate, zoledronate and risedronate are more effective than simple bisphosphonates such as etidronate and tiludronate at suppressing bone turnover in PDB, but their effects on pain are similar. Although bisphosphonates suppress bone turnover in PDB, there is no evidence to show that they alter the natural history or prevent complications. Calcitonin can be used as an alternative but is less convenient to administer and more expensive. Repeated courses of bisphosphonates or calcitonin can be given if symptoms recur. If symptoms do not respond to antiresorptive therapy, it is likely that the pain is due to a complication of the disease and this should be managed according to the principles described.
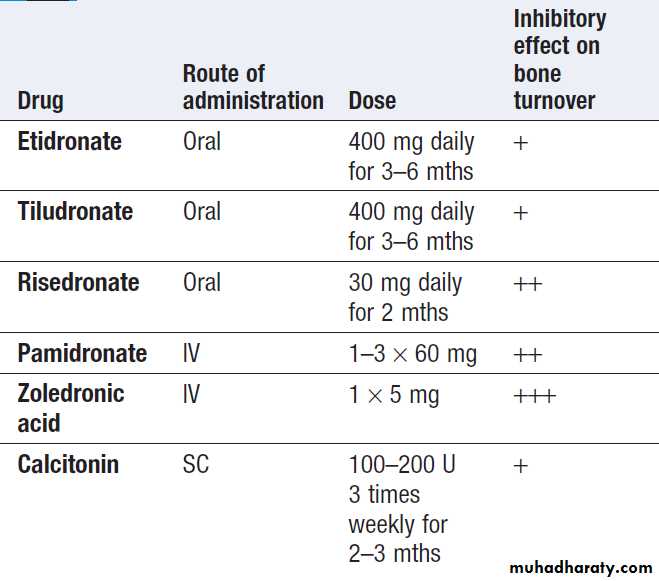
Medical management of Paget’s disease
Other bone diseasesReflex sympathetic dystrophy syndrome
Reflex sympathetic dystrophy syndrome (RSDS), or
algodystrophy, presents with gradual onset of severe
pain, swelling and local tenderness, usually affecting a
limb extremity. It is characterised by localised osteoporosis
of the affected limb and evidence of regional
autonomic dysfunction, such as abnormal sweating,
colour and temperature change. It is commonly triggered
by fracture, occurring in up to 25% of patients
with Colles fracture. It can also associate with soft tissue
injury, pregnancy and intercurrent illness, or can
develop spontaneously.
The cause is unknown but overactivity of the sympathetic nervous system is thought to be responsible for many of its features. The diagnosis is clinical but supported by patchy
osteoporosis of the affected bone on X-ray, by a local
increase in isotope uptake on bone scanning (Fig. 25.58),
or by marrow oedema on MRI. The differential diagnosis
includes infection and malignancy.
Usually, the diagnosis of RSD is clear on clinical grounds and by the absence of an acute phase response or other systemic features of malignancy, but a biopsy of the affected site can be undertaken if necessary and typically shows local osteopenia only.
The aims of treatment are to control pain and encourage
mobilisation. Analgesics, NSAID, antineuropathicagents, calcitonin, corticosteroids, β-adrenoceptor antagonists (β-blockers), sympathectomy and bisphosphonates have all been tried, but none is particularly effective. Although some cases resolve with time, many have persistent symptoms and fail to regain normal function.

Reflex sympathetic dystrophy. Isotope bone scan showing
increased uptake in femoral condyle.Osteonecrosis
Osteonecrosis describes death of bone due to impairmentof its blood supply. The most commonly affected sites are the femoral head, humeral head and femoral
condyles. In some cases, the condition occurs as the
result of direct trauma that interrupts the blood supply
to the affected bone. This is the reason for osteonecrosis
of the femoral head in patients with subtrochanteric
fractures of the femoral neck, and patients with thrombophilia and haemoglobinopathies such as sickle cell
disease. Other important predisposing factors include
high-dose corticosteroid treatment, alcohol excess, SLE
and radiotherapy, but in many of these conditions, the
pathophysiology is poorly understood.
The presentation is with pain localised to the affected site, which is exacerbated by weight-bearing. The diagnosis can be confirmed by MRI, which shows evidence of subchondral necrotic bone and bone marrow oedema. X-rays are normal in the early stages but later may show evidence of osteosclerosis and deformity of the affected bone.
There is no specific treatment.
Management should focus on controlling pain and encouraging mobilization.
Symptoms often improve spontaneously with
time but joint replacement may be required in patients
who have persisting pain in association with significant
structural damage to the affected joint.
Scheuermann’s osteochondritis
This disorder predominantly affects adolescent boys,who develop a dorsal kyphosis in association with
irregular radiographic ossification of the vertebral end
plates. It has a strong genetic component and may be
inherited in an autosomal dominant manner. Most
patients are asymptomatic but back pain, aggravated
by exercise and relieved by rest, may occur. Excessive
exercise and heavy manual labour before epiphyseal
fusion has occurred may aggravate symptoms. Management consists of advice to avoid excessive activity,
and protective postural exercises.
Rarely, corrective surgery may be required if there is severe deformity.
During adulthood, Scheuermann’s osteochondritis cansometimes be complicated by the development of
secondary OA, which may cause back pain. Occasionally,
the vertebral deformity and kyphosis can be mistaken
for osteoporotic vertebral fractures during adult
life, but this can be excluded by DEXA examination,
which typically shows normal or raised BMD values in
Scheuermann’s disease.
Osteogenesis imperfecta
Osteogenesis imperfecta (OI) is the name given to a
group of disorders characterised by severe osteoporosis
and multiple fractures in infancy and childhood. Most
cases are caused by mutations in the COL1A1 and
COL1A2 genes, which encode the proteins that make up
type I collagen. This results either in reduced collagen
production (in mild OI) or in formation of abnormal
collagen chains that are rapidly degraded (in severe OI).
Most patients have dominant inheritance but recessive
forms have been described, caused by mutations in the
CRTAP and LEPRE genes, which are involved in posttranslational modification of collagen.
Severity varies from neonatal lethal (type II),
through very severe with multiple fractures in infancyand childhood (types III and IV), to mild (type I), in
which affected patients typically have blue sclerae. The
diagnosis of OI is usually obvious clinically. In childhood,
the disease can be mistaken for non-accidental
injury and in adulthood for osteoporosis. In such cases,
genetic testing can be of diagnostic value.
Treatment is multidisciplinary, involving surgical reduction and fixation of fractures and correction of limb deformities, and physiotherapy and occupational therapy for rehabilitation of patients with bone deformity. Bisphosphonates are widely used, especially intravenous pamidronate in children, but there is limited
evidence that this prevents fractures or deformity.
Osteopetrosis
Is a rare group of inherited diseases caused by failure of osteoclast function. Presentation is highly variable, ranging from a lethal disorder that presents with bone marrow failure in infancy to a milder and sometimes asymptomatic form that presents in adulthood. Severe osteopetrosis is inherited in an autosomal recessive manner and presents with failure to thrive, delayed dentition, cranial nerve palsies (due to absent cranial foramina), blindness, anaemia and recurrent infections due to bone marrow failure. The adultonset type (Albers–Schِnberg disease) shows autosomal dominant inheritance and presents with bone pain, cranial nerve palsies, osteomyelitis, OA or fracture, or
is sometimes detected as an incidental radiographic finding.
The responsible mutations either affect the
genes that regulate osteoclast differentiation (RANK,RANKL), causing ‘osteoclast-poor’ osteopetrosis, or
affect the genes involved in bone resorption, causing
‘osteoclast-rich’ osteopetrosis. These include mutations
in the TCIRG1 gene, which encodes a component of the
osteoclast proton pump, and mutations in the CLCN7
gene, which encodes the osteoclast chloride pump.
Management is difficult. IFN-γ treatment can improve
blood counts and reduce frequency of infections, but in
severe cases haematopoietic stem cell transplantation is
required to provide a source of osteoclasts that resorb
bone normally.
BONE AND JOINT TUMOURS
Primary tumours of bones and joints are rare, have a peak incidence in childhood and adolescence, and can be benign or malignant . Paget’s disease of bone accounts for most cases of osteosarcomaoccurring above the age of 40.
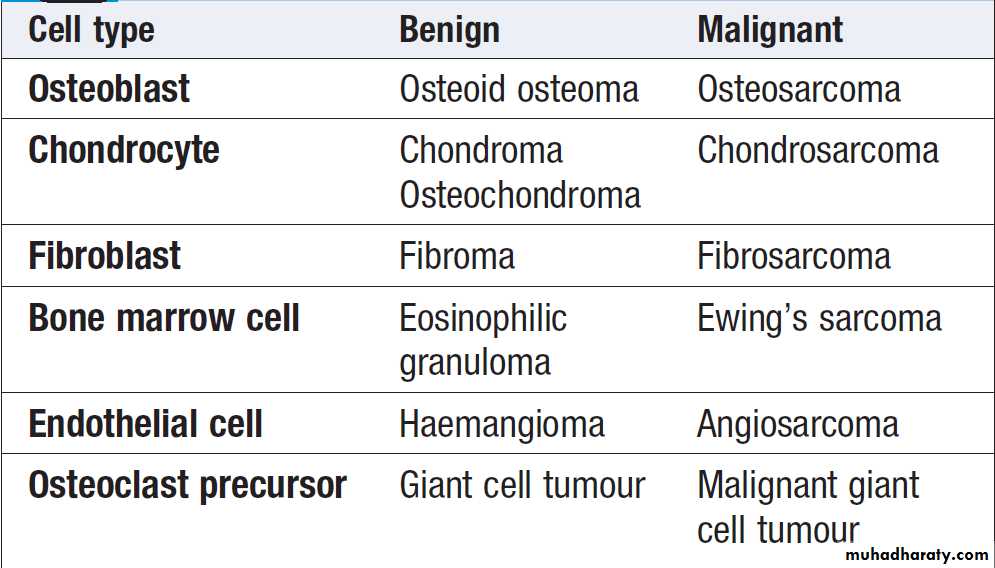
Primary tumours of the musculoskeletal system
OsteosarcomaPresents with local pain, swelling and tenderness. X-rays may show expansion of the bone with a surrounding soft tissue mass, often containing islands of calcification, but further evaluation by MRI or CT is necessary to determine the extent of tumour. The diagnosis can be confirmed by biopsy but this should be done after referral to a specialist team. Treatment depends on histological type but generally involves surgical removal of the tumour, followed by chemotherapy and radiotherapy. The prognosis is excellent with benign tumours and also generally good in cases that present in childhood and adolescence. The prognosis is poor in elderly patients with osteosarcoma related to Paget’s disease of bone.
Metastatic bone disease
Metastatic bone disease may present in a variety ofways: with localised or generalised progressive bone
pain, generalised regional pain, symptoms of spinal cord
compression, or acute pain due to pathological fracture.
Systemic features, such as weight loss and anorexia, and
symptoms referable to the primary tumour are often
present. The tumours that most commonly metastasise
to bone are myeloma and those of bronchus, breast,
prostate, kidney and thyroid.
RHEUMATOLOGICAL INVOLVEMENT IN
OTHER DISEASESMany systemic diseases can affect the locomotor system, and many drugs may cause adverse locomotor effects.
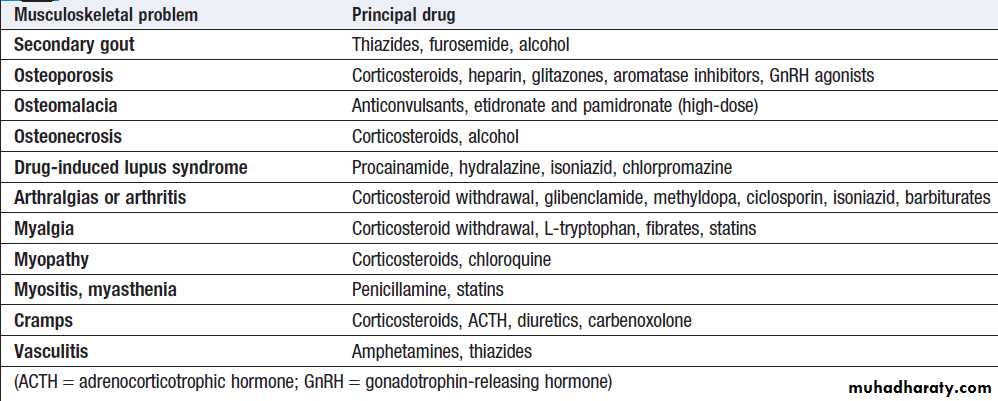
Drug-induced effects on the musculoskeletal system
Malignant diseaseMalignant disease can cause a variety of non-metastatic
musculoskeletal problems . One of the most striking is hypertrophic pulmonary osteoarthropathy, characterised by clubbing and painful swelling of the limbs, periosteal new bone formation and arthralgia/ arthritis. The most common causes are bronchial carcinoma and mesothelioma , but the condition can be inherited when it is caused by inactivating mutations in the HPGD gene, which is responsible for degradation of PGE2, suggesting that over-production of prostaglandins may generally play a causal role in clubbing. Bone scans show increased periosteal uptake before new bone is apparent on X-ray. The course follows that of the underlying malignancy and resolves if this is cured.

Rheumatological manifestations of malignancy
Endocrine diseaseHypothyroidism may present with carpal tunnel syndrome, or rarely with very painful, symmetrical proximal myopathy with muscle hypertrophy. Both resolve with thyroxine replacement. Hyperparathyroidism predisposes to calcium pyrophosphate dihydrate deposition disease and to calcific
periarthritis, especially in patients with renal disease. Diabetes mellitus commonly causes diabetic cheiroarthropathy characterised by tightening of skin
and periarticular structures, causing flexion deformities
of the fingers that may be painful.
Diabetic osteopathy presents as forefoot pain with radiographic progression from osteopenia to complete osteolysis of the phalanges and metatarsals.
Diabetes also predisposes to adhesive capsulitis, Dupuytren’s contracture, septic arthritis and Charcot’s joints.
Acromegaly can be associated with mechanical
back pain, with normal or excessive (not restricted)
movement; carpal tunnel syndrome; Raynaud’s syndrome;
and an arthropathy (50%). The arthropathy
mainly affects the large joints and has clinical similarities
to OA but with normal or an increased range of movement.
X-rays may show widening of joint spaces, squaring
of bone ends, generalised osteopenia and tufting of
terminal phalanges. It does not improve with treatment
of the acromegaly.
Haematological disease
Haemochromatosis is complicated by an arthropathy in about 50% of cases. It typically presents between the ages of 40 and 50, and may predate other features of the disease. The small joints of the hands andwrists are typically affected but the hips, shoulders and
knees may also be involved. The X-ray changes resemble
OA but cysts are often multiple and prominent, with
little osteophyte formation. Involvement of the radiocarpal
and MCP joints may occur, which is unusual in
primary OA, and about 30% have calcium pyrophosphate
dihydrate deposition disease and/or pseudogout.
Treatment of the haemochromatosis does not influence
the arthropathy, and management of the arthropathy is
as described for OA.
Haemophilia can be complicated by haemarthrosis, which, if recurrent, can result in the development of secondary osteoarthritis.
Sickle-cell disease may be complicated by bone pain, osteonecrosis and osteomyelitis.
Thalassaemia may be complicated by bone deformity, especially affecting the craniofacial bones, and by osteoporosis.
Neurological disease
Neurological disease may result in rapidly destructivearthritis of joints, first described by Charcot in association
with syphilis. The cause is incompletely understood
but may involve repetitive trauma as the result of
sensory loss and altered blood flow secondary to
impaired sympathetic nervous system control. The main
predisposing diseases and sites of involvement are:
• diabetic neuropathy (hindfoot)
• syringomyelia (shoulder, elbow, wrist)
• leprosy (hands, feet)
• tabes dorsalis (knees, spine).
The presentation is with subacute or chronic monoarthritis.
Pain can occur, especially at the onset, butonce the joint is severely deranged, pain is often minimal
and signs become disproportionately greater than
symptoms. The joint is often grossly swollen, with
effusion, crepitus, marked instability and deformity,
but usually no increased warmth. X-rays show disorganization of normal joint architecture and often multiple loose bodies and either no (atrophic) or gross (hypertrophic) new bone formation. Management
principally involves orthoses and occasionally arthrodesis.
MISCELLANEOUS CONDITIONS
Spondylolysis and spondylolisthesis
Spondylolysis describes a break in the integrity of the
neural arch. The principal cause is an acquired defect in
pars interarticularis due to a fracture, mainly seen in
gymnasts, dancers and runners, in whom it is an important
cause of back pain. Spondylolisthesis describes the
condition where a defect causes slippage of a vertebra
on the one below. This may be congenital, post-traumatic
or degenerative. Rarely, it can result from metastatic
destruction of the posterior elements. Uncomplicated
spondylolysis does not cause symptoms but spondylolisthesis can cause low back pain aggravated by standing and walking.
Occasionally, symptoms of nerve root or spinal compression may occur. The diagnosis can be
made on lateral X-rays of the lumbar spine but MRI maybe required if there is neurological involvement. Advice
on posture and muscle-strengthening exercises is
required in mild cases. Surgical fusion is indicated for
severe and recurrent low back pain.
Surgical decompression is mandatory prior to fusion in patients with significant lumbar stenosis or symptoms of cauda equine compression.
Diffuse idiopathic skeletal hyperostosis
Diffuse idiopathic skeletal hyperostosis (DISH) is a
common disorder, affecting 10% of men and 8% of
women over the age of 65, and associated with obesity, hypertension and type 2 diabetes mellitus. It is characterised by florid new bone formation along the anterolateral aspect of at least four contiguous vertebral bodies . DISH is distinguished from lumbar spondylosis
by the absence of disc space narrowing and marginal
vertebral body sclerosis, and from ankylosing spondylitis by the absence of sacroiliitis or apophyseal joint fusion. It is usually an asymptomatic radiographic finding but can cause back pain or pain at peripheral sites, such as the heel in association with calcaneal spur formation.
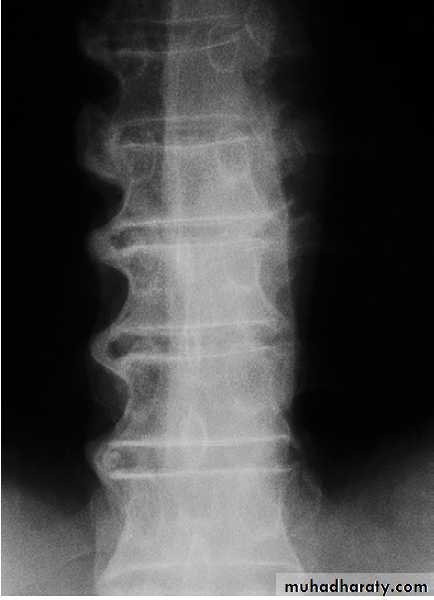
Diffuse idiopathic skeletal hyperostosis (DISH).
Antero-posterior X-ray of thoracic spine showing right-sided, flowing newbone joining more than four contiguous vertebrae. The disc spaces are
preserved.
Pigmented villonodular synovitis
Pigmented villonodular synovitis is an uncommonproliferative disorder of synovium, which typically
affects young adults. It is caused by a somatic chromosomal translocation in synovial cells that places the
CSF1 gene downstream of the COL6A3 gene promoter.
The result is local over-production of macrophage
colony-stimulating factor (M-CSF), which causes accumulation of macrophages in the joint.
The presentation is with joint swelling, limitation of movement and local discomfort. The diagnosis can be confirmed by MRI or synovial biopsy. Treatment is by surgical or radiation synovectomy.
Joint hypermobility
Hypermobility is a relatively uncommon disorder associated with joint laxity.
It may be primary or secondary to inherited diseases, such as Marfan’s syndrome , Ehlers–Danlos syndrome and osteogenesis imperfecta .
Benign joint hypermobility syndrome is diagnosed clinically when the modified Beighton score is 4 or above in the presence of arthralgia in four or more joints .
There is no specific treatment.
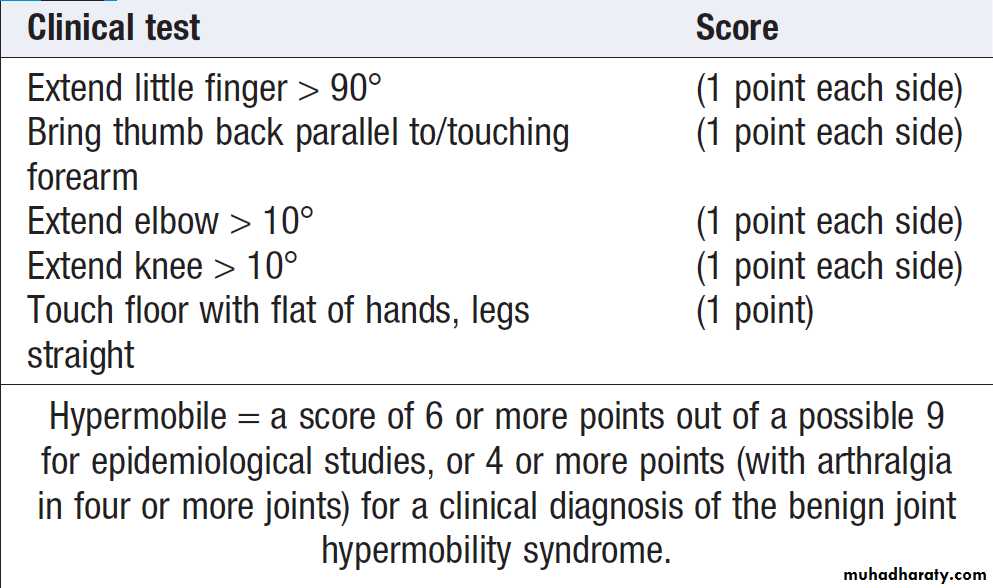
Modified Beighton score for joint hypermobility
Dupuytren’s contractureResults from fibrosis and contracture of the superficial palmar fascia of the hands. The patient is unable to extend the fingers fully and there is puckering of the skin with palpable nodules. The ring and little fingers are usually the first and worst affected. It is usually painless.
It is age-related, usually bilateral and more common in men. There is a strong genetic component and sometimes may be familial, with dominant inheritance. It can be associated with plantar fibromatosis, Peyronie’s disease, alcohol misuse and chronic vibration injury. It is very slowly progressive. Often no treatment is required, but it can be treated medically by local injections
of collagenase or fasciotomy if symptoms are troublesome.
Carpal tunnel syndrome
This is a common nerve entrapment syndrome causedby compression of the median nerve at the wrist. It
presents with numbness, tingling and pain in a median
nerve distribution . The most common causes
are hypothyroidism, diabetes mellitus, RA, obesity and
pregnancy, especially in the third trimester. In some
patients no underlying cause may be identified. Carpal
tunnel syndrome often responds to treatment of the
underlying condition, but other options include local
steroid injections and surgical decompression.
Trigger finger
This occurs as the result of stenosing tenosynovitis in the
flexor tendon sheath, with intermittent locking of the
finger in flexion. It can arise spontaneously or in association with inflammatory diseases like RA. Symptoms
usually respond to local steroid injections but surgical
decompression is occasionally required.
Periodic fever syndromes
These are a group of rare inherited disorders that presentwith intermittent attacks of fever, rash, arthralgia and
myalgia. They usually occur in childhood but may
present for the first time in adulthood.
Anterior tibial compartment syndrome
This is characterised by severe pain in the front of
the lower leg, aggravated by exercise and relieved by
rest. Symptoms result from fascial compression of the
muscles in the anterior tibial compartment and may
be associated with foot drop.
Treatment is by surgical decompression.