1
Fifth stage
Pediatric
Lec. 3
.د
انور
12/4/2017
Thalassemias
Definition
Thalassemias are hereditary hemolytic anemias characterized by decreased or absent
synthesis of one or more globin subunits of the Hb molecule.
α-Thalassemia results from reduced synthesis of α-globin chains.
β-thalassemia results from reduced synthesis of β-globin chains.
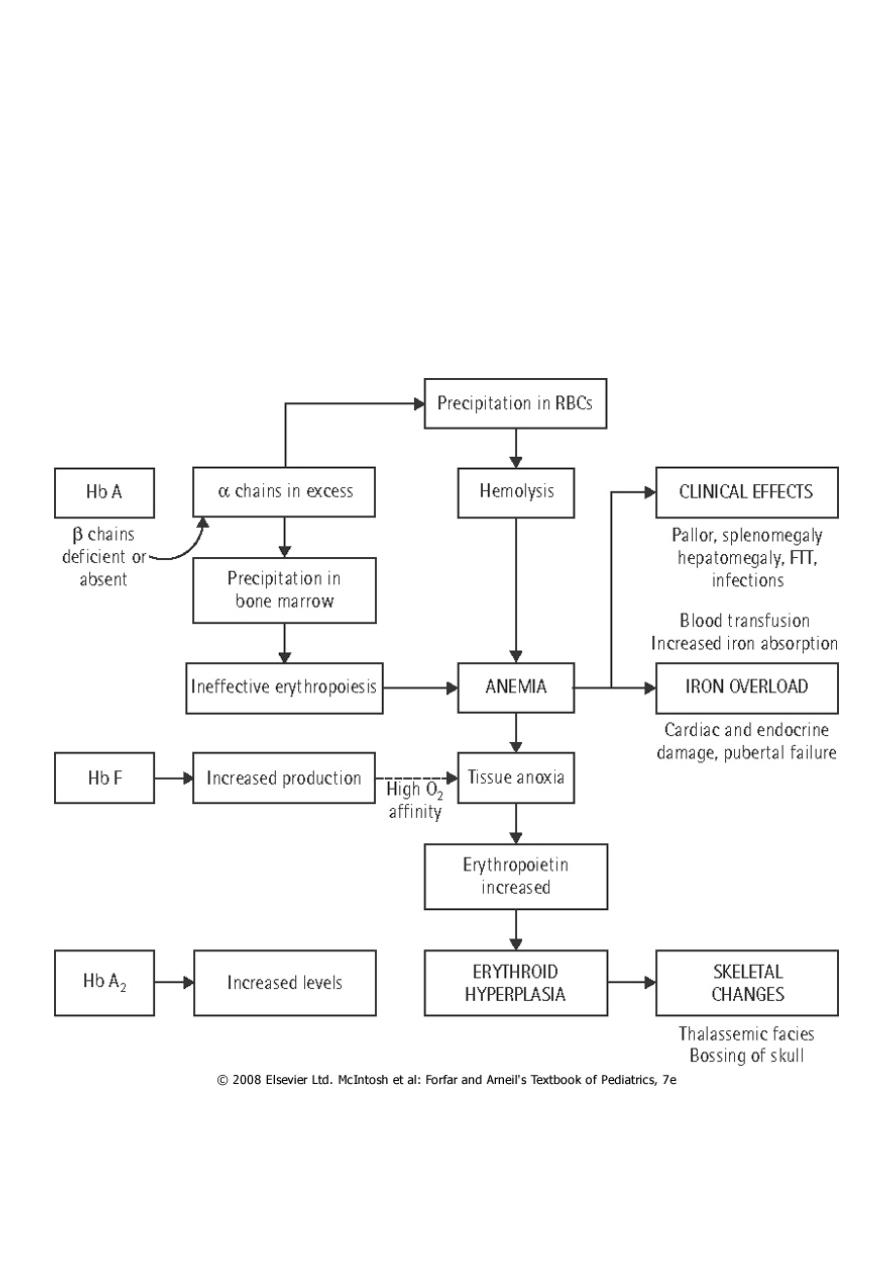
An imbalance in globin chain production is a hazard to the RBC because excess unpaired
globin chains produce insoluble tetramers that precipitate, causing membrane damage.
This makes RBCs susceptible to destruction within the reticuloendothelial system of the BM
(resulting in ineffective erythropoiesis) as well as the RES of the liver and spleen (resulting
in hemolytic anemia).
2
α-Thalassemias
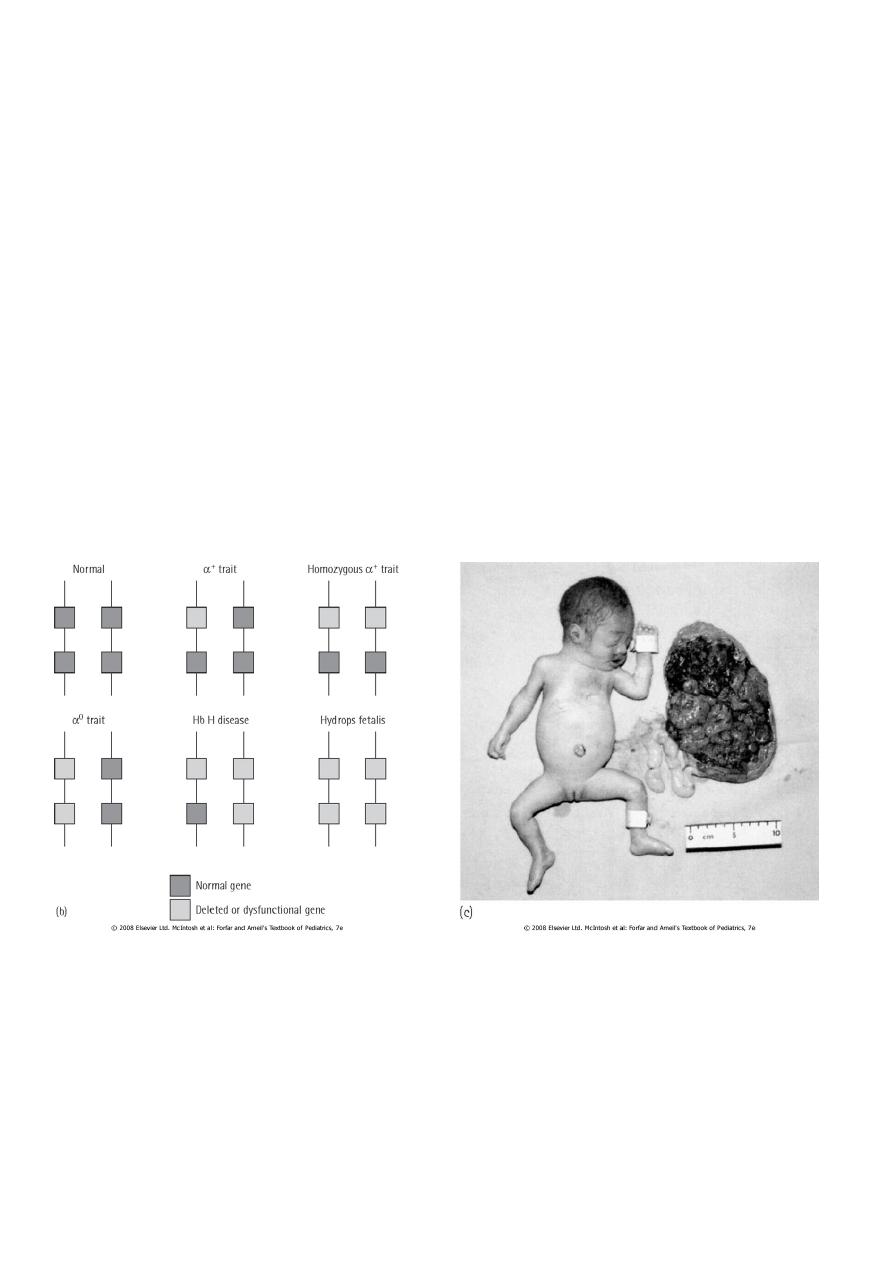
α-Thalassemias are usually the result of gene deletion.
α-Thalassemia variants are found most often in populations of African or East Asian
ancestry.
Normally there are four α-globin genes; clinical manifestations of α-thalassemia variants
reflect the number of genes affected
1. α-thalassemia major: 4 genes deleted (Hb Bart ) , Hydrops fetalis /death in utero .
2. Hemoglobin H disease:3 genes deleted, (Hb H ), baby born with severe anemia, mild
jaundice & splenomegaly
3. α-Thalassemia minor: 2 genes deleted, Mild anemia.
4. Silent carrier: one gene deleted, No anemia

3
β-Thalassemias
The clinical phenotype of β-thalassemia is related to the degree of globin chain imbalance.
1. Heterozygous β-thalassemia (β-thalassemia minor).
2. Homozygous β-thalassemia (β-thalassemia major, Cooley anemia, and intermedia).
Heterozygous β-thalassemia (β-thalassemia minor)
Clinical features
1. Mild anemia (Hb about 10 gm/dl)
2. Normal growth and development.
3. Blood film: Hypochromia, microcytosis, and anisocytosis.
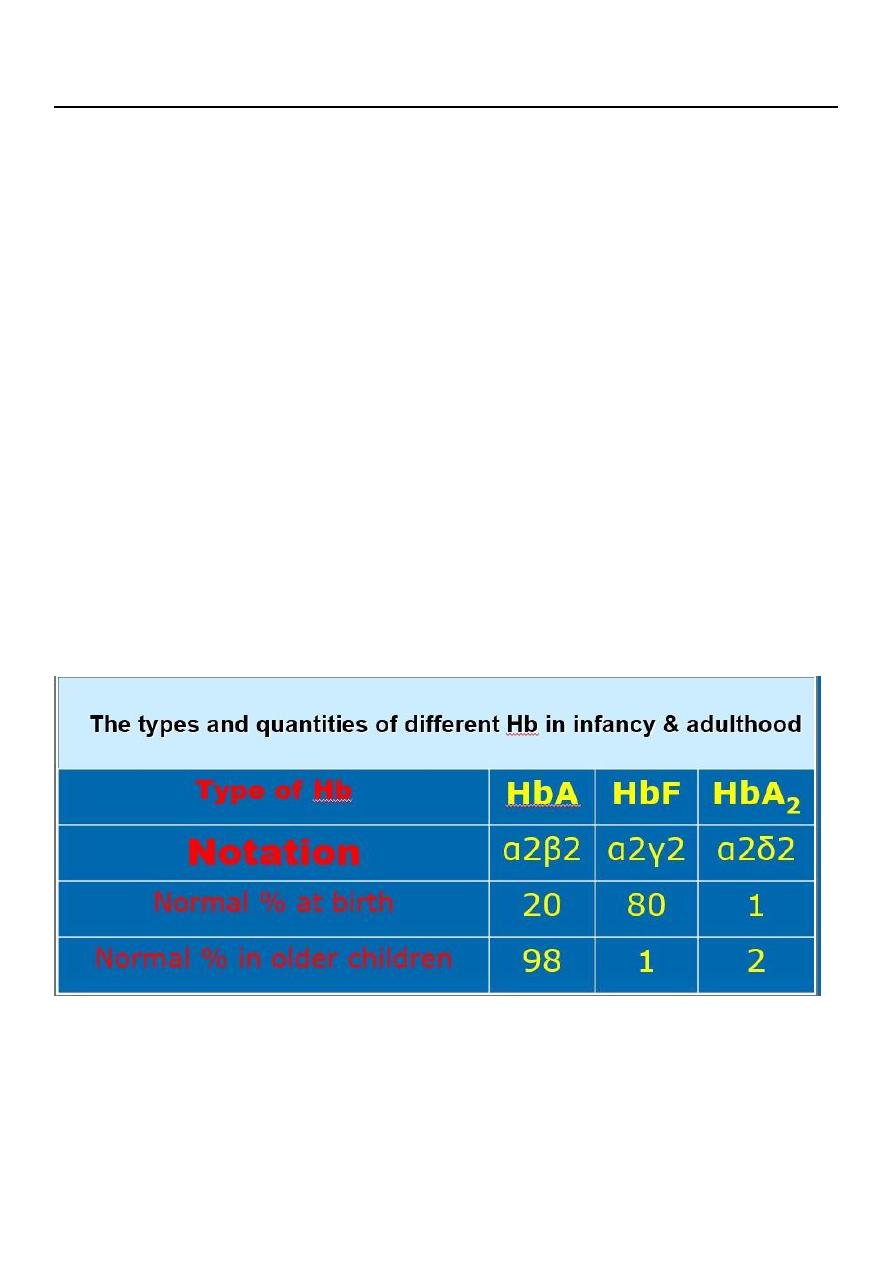
4. Hb electrophoresis shows elevation of the Hb A2 level and, sometimes, elevation of the
Hb F level.
Therapy
No treatment is necessary.
▪ It is important, however, that thalassemia minor is distinguished from ID to prevent
inappropriate therapy with medicinal iron.
▪ Folic acid may be given.
▪ Genetic counseling is also important.
Homozygous β-thalassemia
Homozygous β-thalassemia (β-thalassemia major, Cooley anemia, and intermedia). Patients
who have this form of anemia are usually of Mediterranean background.
Defect. Molecular defects range from complete absence of β-globin synthesis (genotype
β0/β0) to partial reduction in the gene product from the affected locus (genotype β+/β+).
Clinical features: beginning in the middle of the first year of life
1- the infant manifests a progressively severe HA & jaundice .
2- marked HSM.
3- FTT
4
4- BM hyperplasia produces characteristic features such as tower skull, frontal bossing,
maxillary hypertrophy with prominent cheekbones, and overbite.
5- Hemochromatosis: Even in the untransfused state, iron overload develops in thalassemic
pt because of hyperabsorption of dietary iron.
The iron load becomes even greater with chronic transfusion therapy. When the BM
storage capacity for iron is exceeded, iron accumulates in parenchymal organs such as the
liver, heart, pancreas, gonads, and skin, producing the complications of hemochromatosis
(“bronzed diabetes”).
Many patients succumb to congestive HF , hypoparathyroidism, hypogonadism , DM , liver
cirrhosis and short stature.
5
Investigations:
1. CBC : hypochromic microcytic anemia, with nucleated RBC and retics count commonly
less than 8% which is inappropraitely low to the degree of anemia due to ineffective
rythropoiesis).
2. Elevated unconjucated bilirubin.
3. On Hb electrophoresis, Hb A is either markedly decreased or totally absent. Of the total
Hb concentration, 30% to 90% is Hb F.
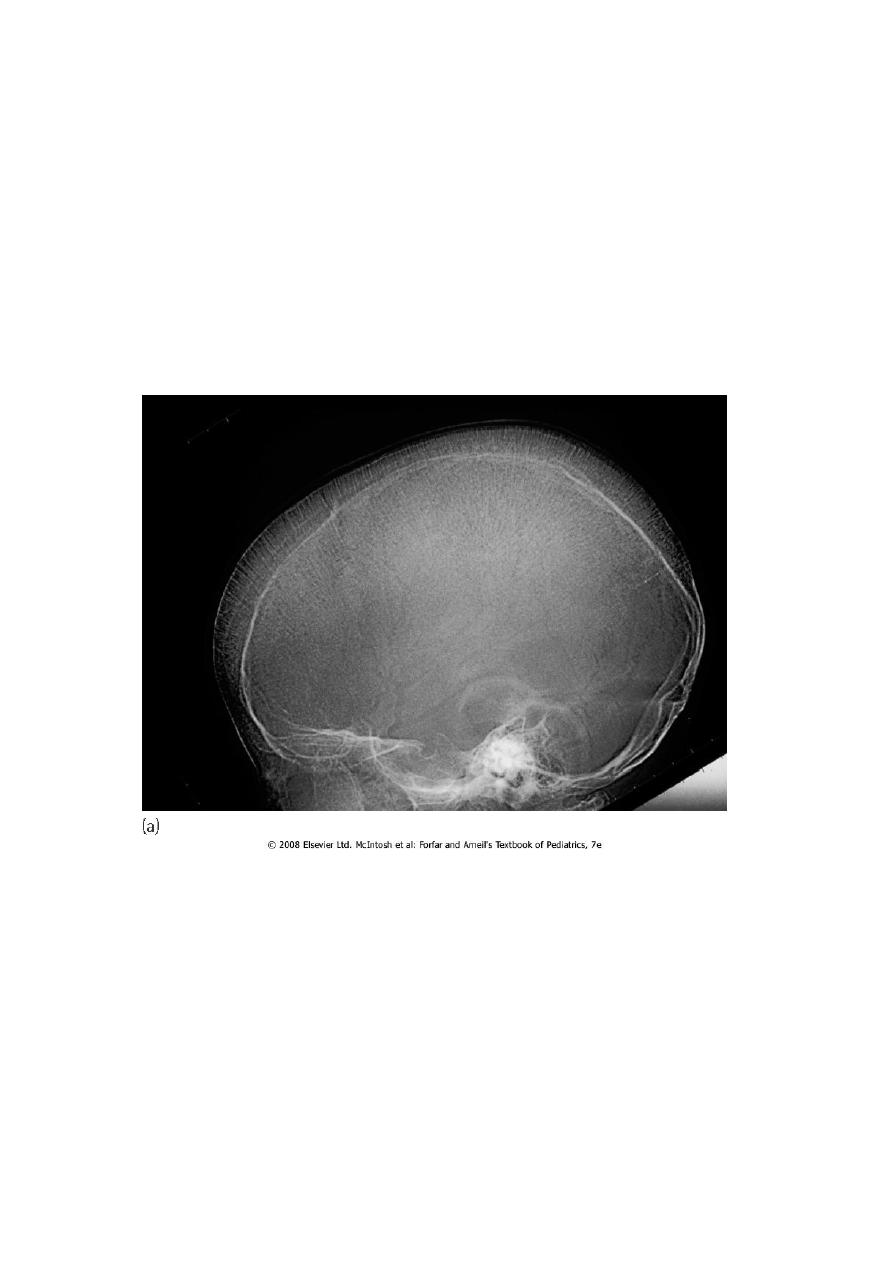
4. BM hyperplasia is seen in bone XR.
5. Elevated S.ferretin & transferrin saturation.
Skull X-ray showing ‘hair on end’ appearance caused by marrow hyperplasia and expansion
Treatment
1- The mainstay of treatment is transfusion with packed RBCs using irradiated CMV –ve
blood, a post transfusion Hb level of 9.5-10 gm/dl is the goal.
2- In an effort to prevent hemochromatosis, pts who receive chronic transfusion regimens
are treated with chelating agents (e.g., deferoxamine, deferiprone) that promote iron
removal from the body through excretion in the urine & stool.
Deferoxamine is given subcutaneously over 10-12 hrs,5-6 days a week.

6
Side effects of deferoxamine includes:
a. ototoxicity.
b. retinal changes.
c. bone dysplasia with truncal shortening.
d. Yersinia bacteria over growth.
3- Splenectomy is usually considered when transfusion requirements exceed 250
mL/kg/year.
4- bone marrow transplantation can cure the patients .
5- Genetic counseling .