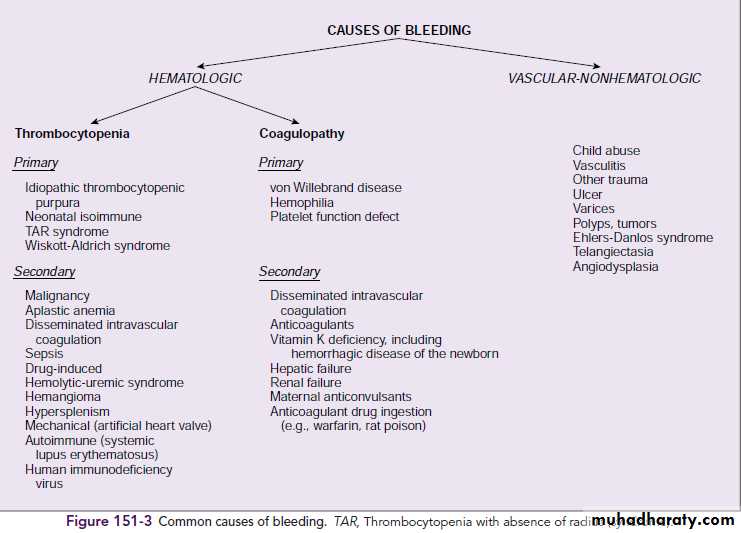
Disorders of Hemostasis
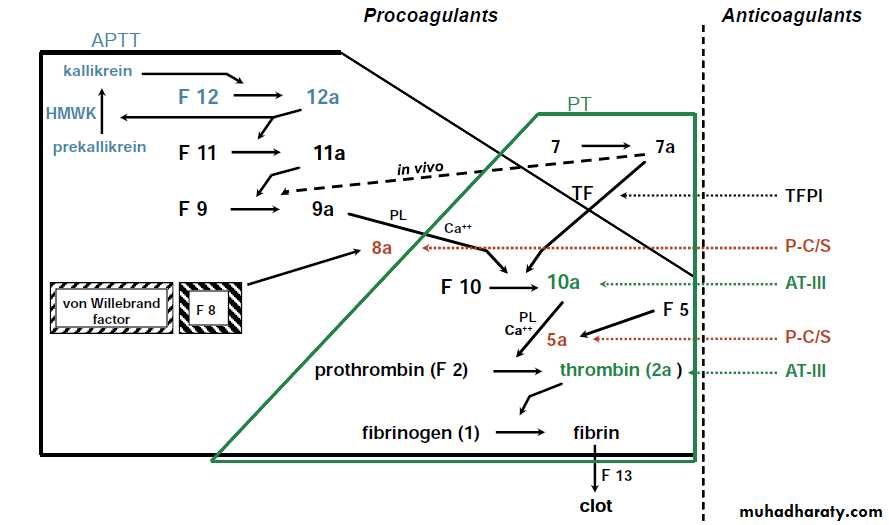
The normal hemostatic mechanism depends upon the integrity of the vessel wall, and in particular the endothelial cells, the presence of normal numbers & function of platelets, and a normal coagulation mechanism.After vascular injury, vasoconstriction occurs and flowing blood comes in contact with the subendothelial matrix .In flowing bd, when exposed to subendothelial matrix proteins, von Willebrand factor (VWF) changes conformation and provides the glue to which the platelet VWF receptor binds. After adherence, plets become activated and release storage granules containing adenosine diphosphate (ADP), thromboxane A2, and other stored proteins.
These trigger the aggregation and recruitment of other plets to form the plet plug. Aggregation involves the interaction of specific receptors on the plet surface with plasma hemostatic proteins, primarily fibrinogen.
Coagulation pathway
PHYSICAL EXAMINATION.
It should focus on whether:• mucocutaneous bleeding
• muscles and joints (deep bleeding).
Pts with defects in platelet–blood vessel wall interaction (VWD or platelet function defects) usually have mucocutaneous bleeding, which may include epistaxis, menorrhagia, petechiae, ecchymoses, occasional hematomas, and less commonly, hematuria and GIT bleeding.
LABORATORY TESTS.
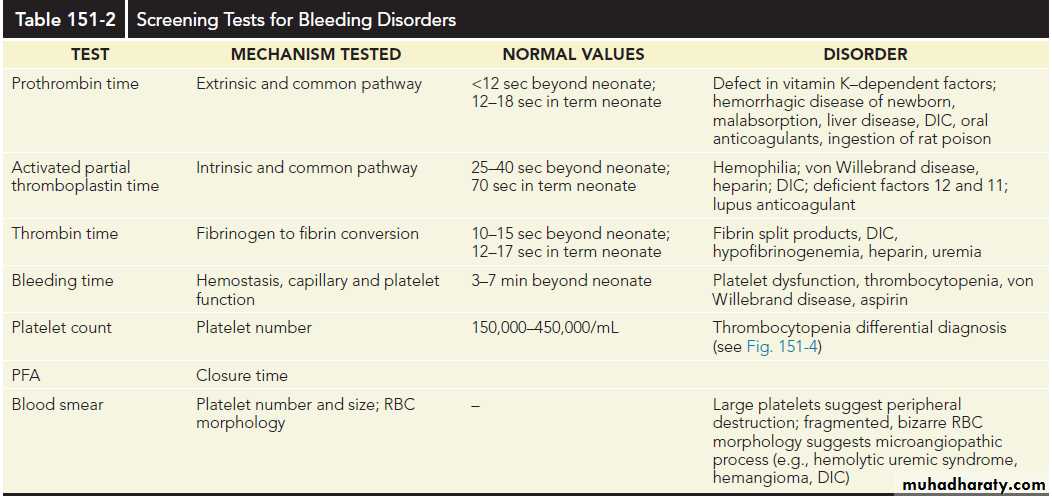
Pts who have a positive bleeding history or who are actively bleeding should have:
platelet count, PT, and partial thromboplastin time (PTT).
If the results are normal, a thrombin time to evaluate fibrinogen function and VWF testing should be considered.
In individuals with abnormal screening test results, further specific factor work-up should be undertaken.
In a pt with an abnormal bleeding history and a positive family history, normal screening tests should not preclude further laboratory evaluation.
Idiopathic Thrombocytopenic Purpura
Etiology. Autoimmune thrombocytopenic purpura of childhood (childhood ITP) is a common disorder that usually follows an acute viral infection. Childhood ITP is caused by an antibody (IgG or IgM) that binds to the plet membrane.The condition results in Fc receptor–mediated splenic destruction of antibody-coated plets. Rarely, ITP may be the presenting symptom of an autoimmune disease, such as SLE.
Clinical Manifestations. Young children typically exhibit ITP 1 to 4 wks after viral illness, with abrupt onset of petechiae, purpura, and epistaxis. The thrombocytopenia usually is severe.
Significant adenopathy or HSM is unusual, and RBC & WBC counts are normal.
Diagnosis.
It usually is based on clinical presentation & plt count & does not often require a BM examination. If atypical findings are noted,however, marrow examination is indicated to rule out an infiltrative disorder (leukemia) or an aplastic process (aplastic anemia). In ITP, an examination of the BM reveals increased megakaryocytes and normal erythroid and myeloid elements.
Treatment and Prognosis.
Therapy is seldom indicated for plt counts > 30,000/mm3.Therapy does not affect the long-term outcome of ITP but is intended to increase the plt count acutely.
For moderate & severe clinical bleeding with severe thrombocytopenia (platelet count <10,000/mm3), therapeutic options include prednisone, 2 to 4 mg/kg/24 hours for 2 weeks or IVIG, 1 g/kg/24 hours for 1 to 2 days.
All of these approaches seem to decrease the rate of clearance of sensitized platelets, rather than decreasing production of antibody. The optimal choice for therapy (if any) is controversial.
Splenectomy is indicated in acute ITP only for ife-threatening bleeding. Approximately 80% of children have a spontaneous resolution of ITP within 6 months after diagnosis.
Serious bleeding, especially intracranial , occurs in fewer than 1% of pts with ITP. There is no evidence that early treatment prevents ICH.
ITP that persists for 6 to 12 ms is classified as chronic ITP. Repeated treatments with IVIG, IV anti-D, or high-dose pulse steroids are effective in delaying the need for splenectomy.
Secondary causes of chronic ITP, especially SLE and HIV infection, should be ruled out. Splenectomy induces a remission in 70% to 80% of childhood chronic ITP cases. The risks of splenectomy (surgery, sepsis from encapsulated bacteria, pulmonary hypertension) must be weighed against the risk of severe bleeding.
Hemophilia
Etiology.Hemophilia A (factor 8 deficiency) occurs in 1 in 5000 males. Hemophilia B (factor 9 deficiency) occurs in approximately 1 in 25,000.
Clinically the two disorders are indistinguishable other than by their therapy .The lack of factor 8 or factor 9 delays the generation of thrombin, which is crucial to forming a normal, functional fibrin clot and solidifying the platelet plug that has formed in areas of vascular injury. The severity of the disorder is determined by the degree of clotting factor deficiency.
Clinical Manifestations.
Pts with < 1% (severe) factor 8 or factor 9 may have spontaneous bleeding or bleeding with minor trauma.Pts with 1% to 5% (moderate ) factor 8 or factor 9 usually require moderate trauma to induce bleeding episodes.
In mild (>5% factor 8 or factor 9), significant trauma is necessary to induce bleeding; spontaneous bleeding does not occur.
Mild hemophilia may go undiagnosed for many yrs, whereas severe hemoph manifests in infancy when the child reaches the toddler stage. In severe hemoph, spontaneous bleeding occurs, usually in muscles or joints (hemarthroses).
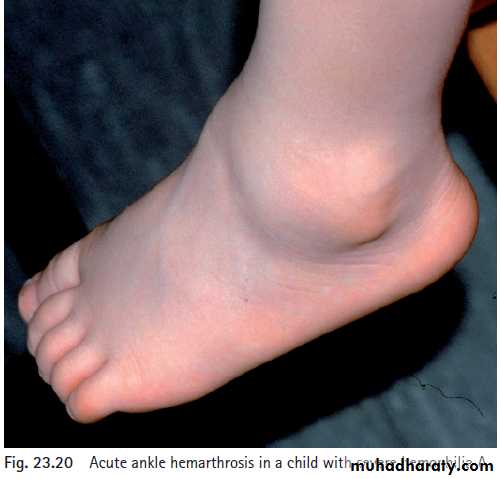
Laboratory Studies.
The diagnosis of hemophilia is based on a prolonged aPTT. In the aPTT, a surface-active agent activates the intrinsic system of coagulation, of which factors 8 and 9 are crucial components.In factor 8 or factor 9 deficiency, the aPTT is quite prolonged but should correct to normal when the patient’s plasma is mixed 1:1 with normal plasma. When an abnormal aPTT is obtained, specific factor assays are needed to make a precise diagnosis to determine the appropriate factor replacement therapy.
Prenatal Dx and carrier Dx are possible using molecular techniques.
Treatment.
Early, appropriate replacement therapy is the hallmark of excellent hemophilia care.
Prophylactic therapy starting in infancy has greatly diminished the likelihood of chronic arthropathy in children with hemophilia. For life-threatening bleeding, levels of 80% to 100% of normal factor 8 or factor 9 are necessary. For mild to moderate bleeding episodes (hemarthroses), a 40% level for factor 8 or a 30% to 40% level for factor 9 is appropriate. The dose can be calculated using the knowledge that 1 U/kg body weight of factor 8 increases the plasma level 2%, whereas 1.5 U/kg of recombinant
factor 9 increases the plasma level 1%:
Dose for F 8 = desired level (%)× weight (kg) × 0.5
Dose for recombinant F 9 = desired level (%) ×weight (kg) × 1.5
Desmopressin acetate is a synthetic vasopressin analog with minimal vasopressor effect. Desmopressin triples or quadruples the initial factor 8 level of a pat with mild or moderate (not severe) hemophilia A, but has no effect on factor 9 levels.
When adequate hemostatic levels can be attained, desmopressin is the Rx of choice for individuals with mild & moderate hemophilia A. Aminocaproic acid is an inhibitor of fibrinolysis that may be useful for oral bleeding.
Patients treated with older factor 8 or 9 concentrates derived from large pools of plasma donors were at high risk for hepatitis B, C, and D and HIV.
Recombinant factor 8 and factor 9 concentrates are safe from virally transmitted illnesses. Acquired immunodeficiency syndrome (AIDS) is the most common cause of death in older hemophilia patients (who received plasmaderived factors). Many older patients also have chronic hepatitis C.
Inhibitors are IgG antibodies directed against transfused factor 8 or factor 9 in congenitally deficient pats.
Inhibitors arise in 15% of severe factor 8 hemophiliacs but are less common in factor 9 hemophiliacs.
They may be high or low titer and show an anamnestic response to treatment. The Rx of bleeding pats with an inhibitor is difficult. For low titer inhibitors, options include continuous factor 8 infusions. For high titer inhibitors, it is usually necessary to administer a product that bypasses the inhibitor, preferably recombinant factor 7a.
von Willebrand Disease
(VWD )Etiology.
VWD is a common disorder (1% of the population) caused by a deficiency of vWF, an adhesive protein that serves two functions: acting as a bridge between subendothelial collagen and platelets and binding & protecting circulating factor 8 from rapid clearance from circulation.
VWD usually is inherited as an autosomal dominant trait and rarely as an autosomal recessive trait. vWF may be either quantitatively deficient (partial = type 1 or absolute = type 3) or qualitatively abnormal (type 2 = dysproteinemia).
Approximately 80% of pts with VWD have classic (type 1 disease (i.e., a mild to moderate deficiency of vWF).
Several other subtypes are clinically important, each requiring different therapy.
Clinical Manifestations.Mucocutaneous bleeding, epistaxis, gingival bleeding, cutaneous bruising, and menorrhagia occur in pts with VWD.
In severe disease, factor 8 deficiency may be profound, and the pt may also have manifestations similar to hemophilia A (hemarthrosis). Findings in classic VWD differ from findings in hemophilia A and B.
Laboratory Testing.
vWF testing involves measurement of the amount of protein, usually measured immunologically as the vWF antigen (vWF:Ag). vWF activity (vWF:Act) is measured functionally in the ristocetin cofactor assay (vWFR:Co), which uses the antibiotic ristocetin to induce vWF to bind to plets.
Treatment.
It depends on the severity of bleeding.Desmopressin is the treatment of choice for most bleeding episodes in pts with type 1 disease and some pts with type 2 disease.
When high levels of vWF are needed but cannot be achieved satisfactorily with desmopressin, Rx with a virally attenuated, vWF-containing concentrate (Humate P) may be appropriate.
The dosage can be calculated as for factor 8 in hemophilia.
Cryoprecipitate should not be used because it is not virally attenuated. Hepatitis B vaccine should be given before the pt is exposed to plasma-derived products. As in all bleeding disorders, aspirin should be avoided.