Chronic myeloid leukaemia( CML);
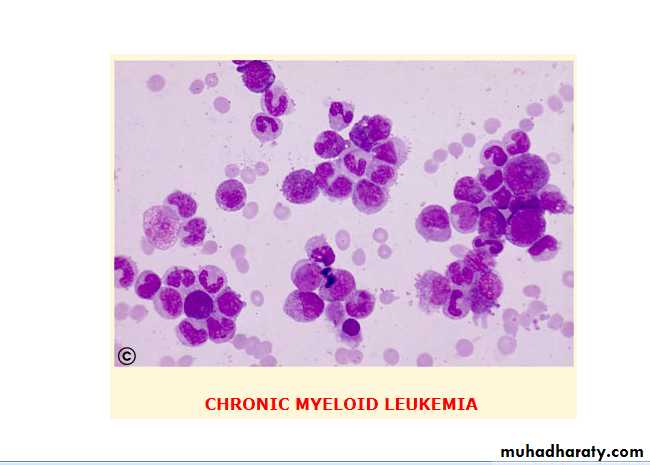
CML is an excessive proliferation with fairly normal maturation. The disease occurs mainly between 30 and 80 years with a peak incidence at 55 years. It is higher in men than in women (1.7:1).
Etiology:
The etiology is unknown however large doses of radiation can induce CML.Cytogenetic and molecular aspects:
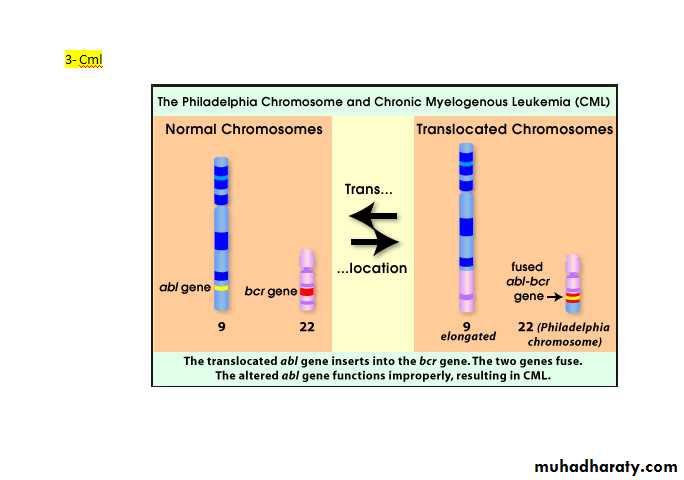
90% of patients with CML have a chromosome abnormality known as the Philadelphia (Ph) chromosome which is due to translocation between chromosome 22 and 9.
The break on chromosome 22 occurs in the breakpoint cluster region (BCR) which form with the fragment from chromosome 9 the Abelson (abl) oncogene, which forms achimeric gene. This chimeric gene codes for protein with tyrosine kinase activity, which plays a causative role in the disease. Some patients with ph-negative also have evidence of the same molecular abnormality.
Natural history:
1- chronic phase in which the disease is responsive to treatment.
2- Accelerated phase in which disease control becomes more difficult.
3- Blast crises in which disease transform into an acute leukaemia (either myeloid 70% or lymphoblastic 30%).
Disease acceleration is defined by the development of increasing degree of anaemia, cytogenetic clonal evolution, or blood or marrow blast between 10-20%, blood or marrow basophils ≥ 20%, or platlets count < 100.000/ML.
Blast crises is defined as acute leukaemia with a blood or marrow blast ≥ 20%.
Clinical features:
1- Generally the presentation is insidious, some patients are diagnosed accidently during routine test.2- Other patients present with fatigue, weight loss, breath-lesness, abdominal discomfort from splenic enlargement, sweating with vague ill health.
3- Less commonly feature related to granulocyte or platlets dysfunction (infection, thrombosis or bleeding) or with leukostatic manifestation due to severe leukocytosis or thrombosis.
Physical finding:
mild to moderate splenomegally, occasionally mild hepatomegally, lymphadenopathy, and extra-medullary tumor are unusual except late in the disease.
Investigation:
1- Normocytic normochronic anaemia.2- Elevated W.B.C count with various degrees of immaturity of the granulocytic series.
3- The mean platlets count is 445 x 109/L.
4- Blast cells in the peripheral circulation usually less than 10%.
5- Leukocyte alkaline phosphatase is characteristically low in CML cells.
6- Serum level of vitamin B12 and B12 binding proteins are generally elevated.
7- Basophilia increased in later stages causing pruritis due to histamine production.
* Blood film shows full range of granulocyte precursor with peaks at the myelocyte and mature granulocyte stage of maturation.
Bone marrow cellularity is increased in patients with CML with blast cell is generally normal or slight elevated. Bone marrow material should be obtained for chromosome analysis.
Management:
No specific therapy is required if the patient is asymptomatic and the leukocyte count not greatly elevated.1- Chemotherapy: hydroxyurea is the most widely used oral agents. 2-4 gm/day is used initially, reduced to maintain the white count in the normal range. However, it does not affect the Philadelphia chromosome frequency.
2- Alpha-interferon given I.M or subcutaneous at 3-9mega-unit daily. Interferon prolong survival in those with significant reduction in ph positive cells.
Severe bone pain and severe weigh loss are reasons for discontinuation.
3- Initab mesylate(Imatinib): this agent is an inhibitor of the BCR-abl. Tyrosine kinase. Over 76% achieving ph-chromosome negatively. It is also very active in interferon resistant cases, in accelerated phase and blast crises.
4- Allogenic or syngeneic bone marrow transplant: this provide the only means of obtaining long term remission in this disease. It is available to those under 55 years of age.
Treatment of the accelerated phase is more difficult. Hydroxyurea (hydroxyl carbamide) can be an effective single agent. Low dose cytarabine can also be tried.
Chronic lymhocytic leukaemia( CLL):
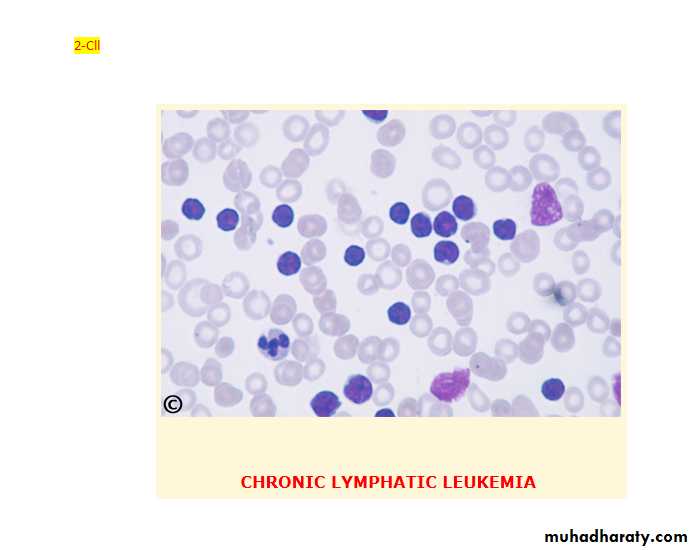
This is the most variely of leukaemia (30% of cases). Male to female ratio is 2:1 with a peak at 65. In this disease B-lymphocyte failed to transform into antibody forming cells in response to antigen. This lead to increasing of immuno-in competent cells and impaired immune function. The B-cells(CD19,CD23) of chronic lymphocytic leukaemia characteristically express a T-cell antigen, CD5.
Clinical features:
1- The onset is very insidious (around 25% of patients are diagnosed incidentally).2- Patients may present with anaemia, painless lymphodenopathy or splenomegally.
3- nfections often occur later in the disease.
Staging of CLL:
1-Clinical stage A 60%: no anaemia or thrombocytopenia and less than three areas of lymphoid enlargement.2-Clinical stage B 30%: no anaemia or thrombocytopenia with three or more involved area of enlargement.
3- Clinical stage C 10%: anaemia and/ or thrombocytopenia, regardless the number of areas of lymphoid enlargement.
Investigations:
1- Mild anaemia. Haemolytic anaemia may occur (warm auto immune type).2- In the majority of patients the leukocyte count is between 50-200 x109/L although it may occasionally be greatly increased. About 95% or more of these cells are mature lymphocytes.
3-Bone marrow examination may be helpful in the diagnosis of cases with a low white count and for prognosis.
4- Chromosome analysis can be helpful (cases with trisomy 12 or 13q abnormalities are associated with a poor prognosis).
5- Estimation of total protein and immunoglobulin level should be undertaken to establish the degree of immune suppression.
In some patients there may be a monoclonal band.
Management:
Treatment depends upon the stage of the disease:1- Clinical stage A: no specific treatment is required.
2- Clinical stage B: chemotherapy with chlorambucil may be initiated (5 mg orally daily over long periods with dose adjustment according to blood counts or intermittent high dose (0.4 mg/kg) every 2 weeks increased by 0.1 mg/kg until the maximum tolerated dose is reached. Local radiotherapy to lymph node may be given if causing discomfort.
3- Clinical stage: correction of anaemia with red cell concentrate. Treatment of bone marrow failure with prednisolon 40 mg daily for 2-4 weeks. More aggressive combination chemotherapy may be beneficial. Fludarabine, a synthetic nucleoside appears to be the most active drug.
Total body irradiation using very small doses spread over 5 weeks in 10 fraction is effective and well tolerated especially by the elderly.
Splenoctomy may be required to treat autoimmune haemolytic anaemia or gross splenic enlargement.