Renal pathology/2
Dr.Shatha Th. Ahmedassistant professor/Ninevah college
PhD pathology/UK
The Nephritic Syndrom
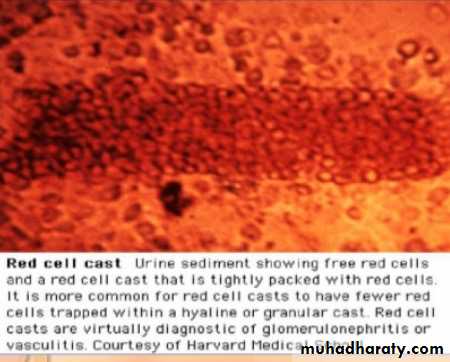
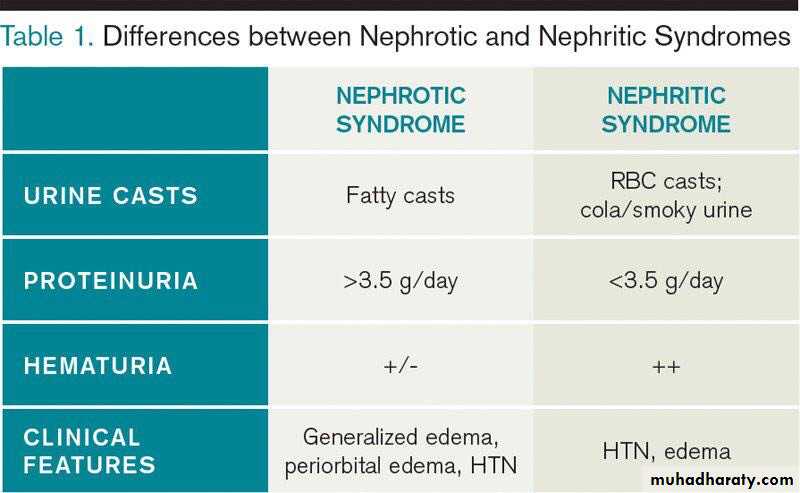
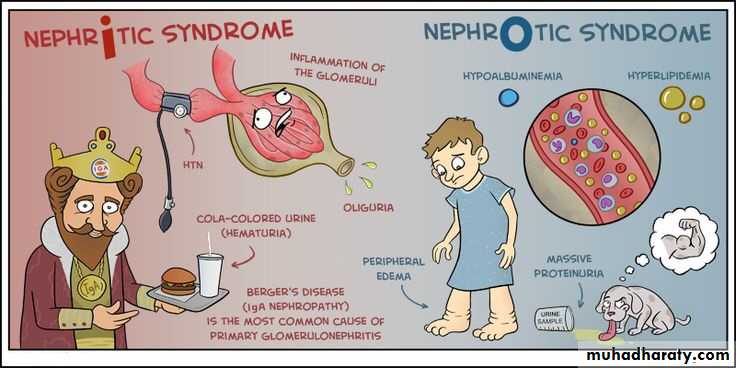
this is a clinical complex, of acute onset, characterised by:1-hematurea
2-oligurea with azotemia
3-hypertension
The lesion that cause nephritic syndrom is inflammatory reaction injures the capillary wall leading to escape of red blood cells,reduced GFR>oligurea, fluid retention and azotemia and hypertension.
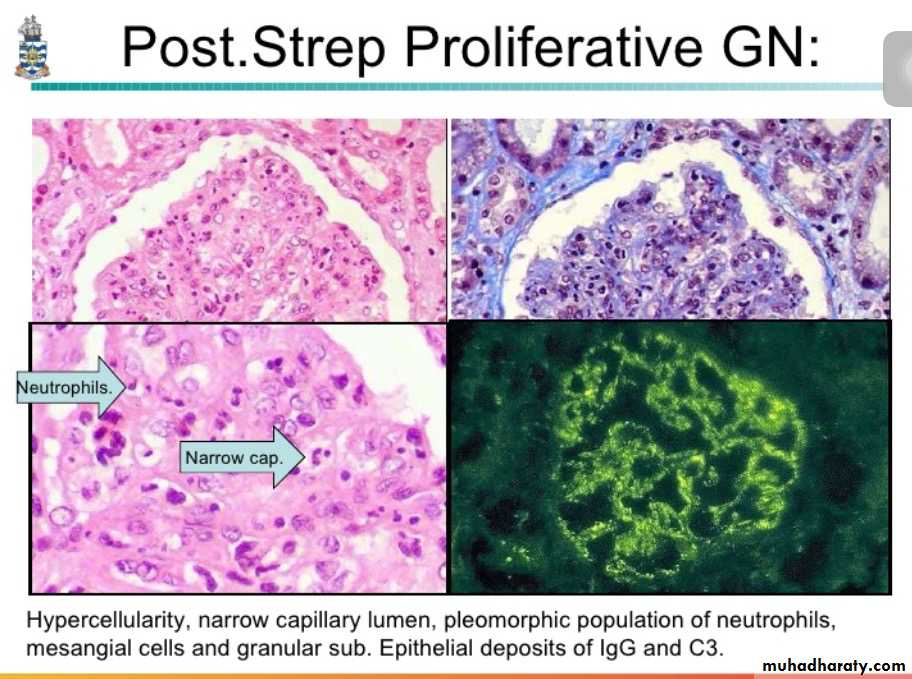
are commonly associated with proliferation of glomerular cells+leukocyte infiltration
causes of Nephritic syndrom
there are 2 causes both are immunologically mediated chx by proliferative changes and inflammation in glomeruli they are1. post infectious GN
2. IgA nephropathy ( Berger's disease)
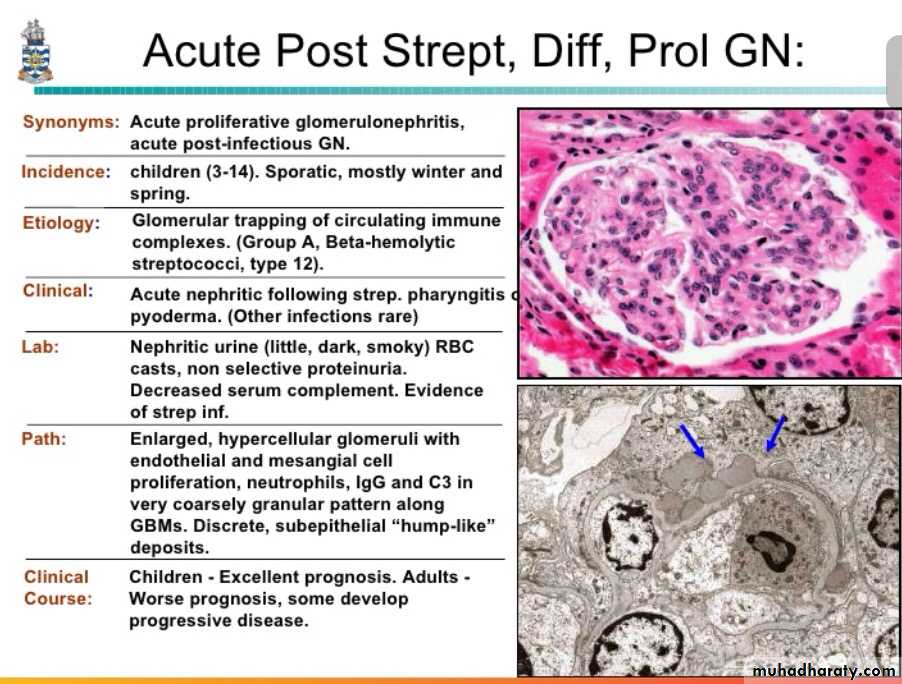
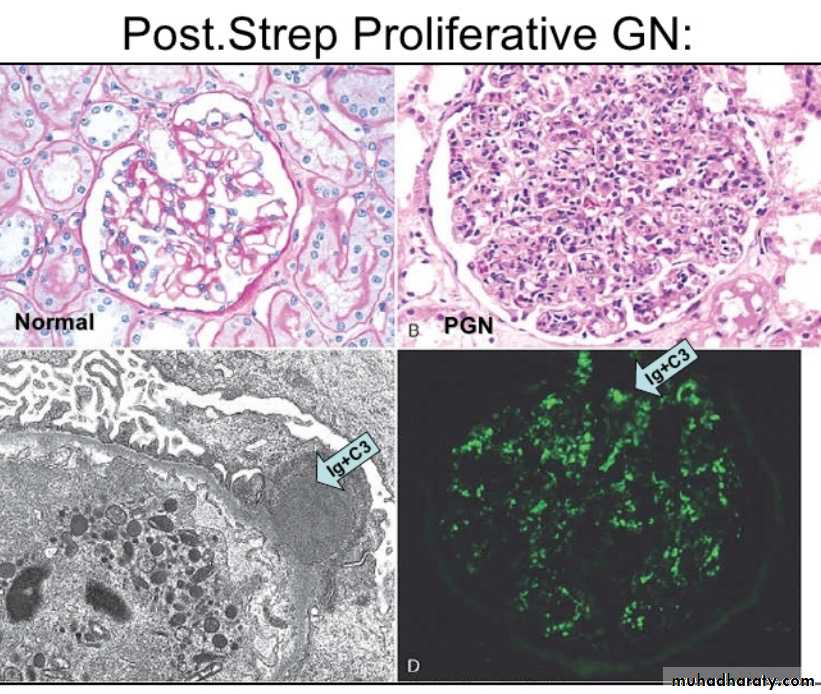
The hypercellularity of post-streptococcal glomerulonephritis is due to increased numbers of epithelial, endothelial, and mesangial cells as well as neutrophils in and around the capillary loops. This disease may follow several weeks after infection with certain strains of group A beta hemolytic streptococci. Patients typically have an elevated anti-streptolysin O (ASO) titer.
2/Rapidly Progressive (Crescentic) Glomerulonephropathy
It is a syndrome associated with severe glomerular injury and is characterized clinically by rapid and progressive loss of renal function, and if untreated, death from renal failure within weeks or months.Classification and pathogenesis
In most of the cases, immunologic injury of the glomeruli is present. Accordingly 3 groups are identified1. Type I RPGN (Anti-GBM AB-Mediated)
* Idiopathic
*Goodpasture Syndrome:characterized by linear IgG deposits along theGBM. In some of these individuals the anti-GBM antibodies also bind to pulmonary alveolar capillary basement membranes to produce the clinical picture of pulmonary hemorrhages associated with renal failure
2. Type II RPGN (Immunecomplex-Mediated) *Idiopathic * Postinfectious * Systemic lupus erythemotosus SLE *Henoch-Schonlein purpura (IgA) and others
3.
Classification and pathogenesis
Type III RPGN (Pauci-Immune) ANCA associated Idiopathic Wegener granulomatosis Microscopic polyarteritis nodosa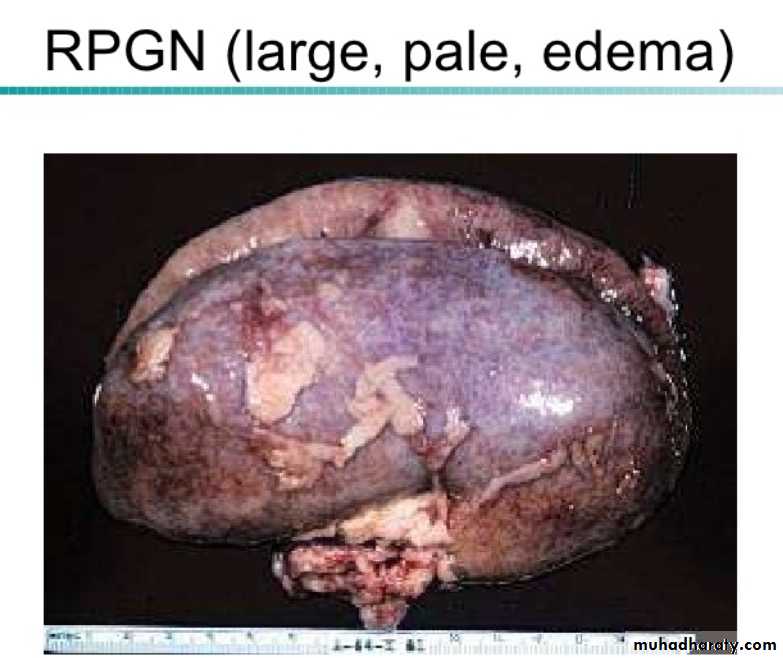
The kidneys are enlarged and pale, often with petechial hemorrhages on the cortical surfaces.
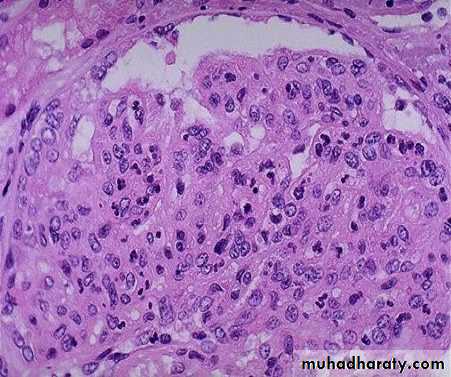
The histologic picture is dominated by the formation of crescents.
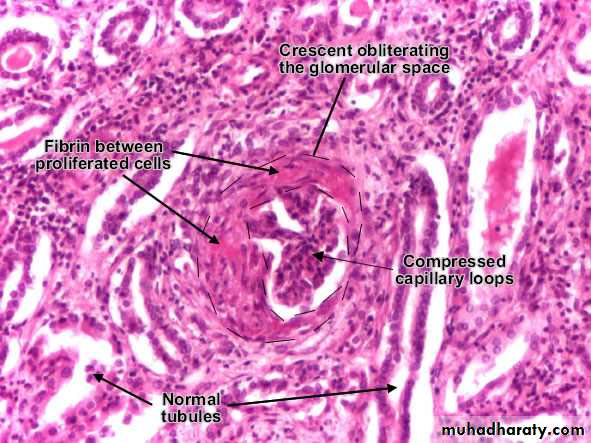
Regardless of the cause, the classic histologic picture is the presence of crescents in most of the glomeruli these are produced by proliferation of the parietal epithelial cells associated with infiltration by monocytes and macrophages
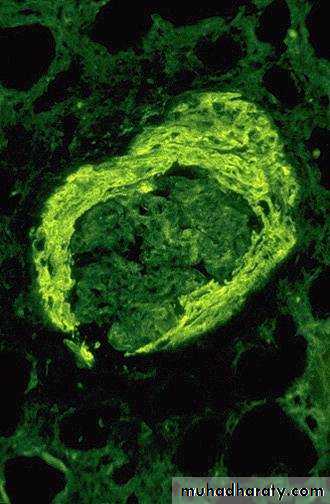
By immunofluorescence microscopy, postinfectious cases exhibit granular immune deposits; Goodpasture syndrome cases show linear deposits while pauci-immune cases have little or no deposits.
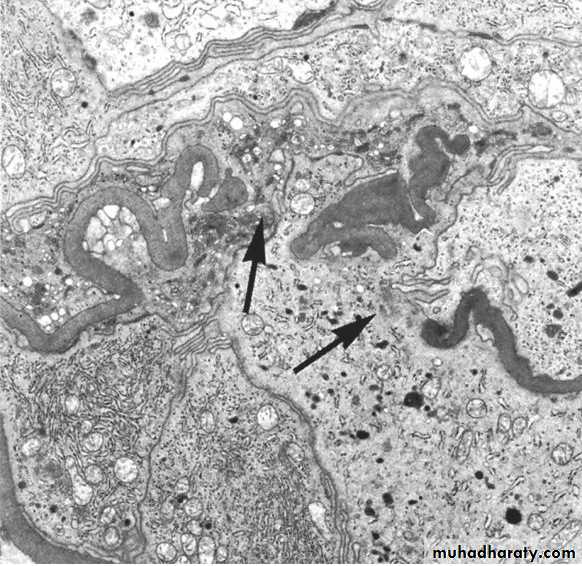
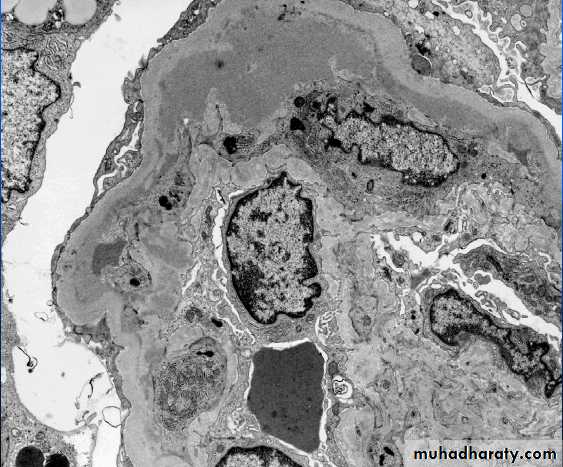
Electron microscopy shows distinct rupture in the basement membrane.
Nephrotic SyndromeCertain glomerular diseases virtually always produce the nephrotic syndrome.
The manifestations include;
1. Massive proteinuria.2. Hypoalbuminemia.
3. Generalized oedema.
4. Hyperlipidemia and lipiduria.
Nephrotic Syndrome-cont


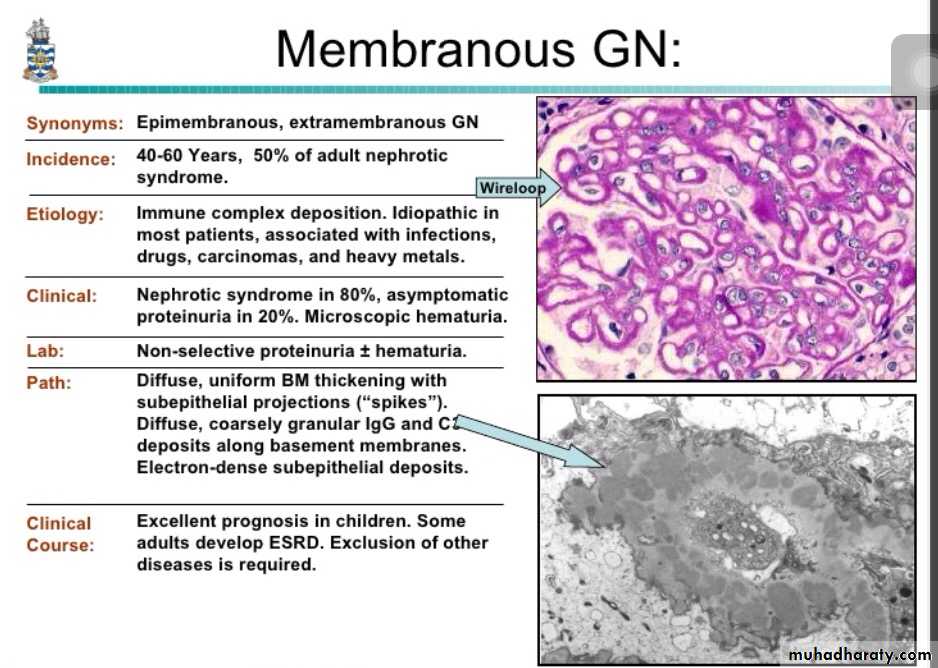
Etiology and pathogenesis,
it is a form of chronic immune-complex-mediated disease.The disease is idiopathic in about 85% of cases, and is called primary MGN. However, secondary forms are identified in association with;
1. Drugs (penicillamine, captopril, NSAIDS).
2. Underlying malignant tumors, particularly bronchogenic carcinoma, carcinoma of colon and melanoma.
3. SLE.
4. Infections (Chronic hepatitis B and C, syphilis, malaria).
5. Autoimmune diseases such as thyroiditis.

By light microscopy, the glomeruli exhibit diffuse thickening of the glomerular capillary wall.
By electron microscopy, there are irregular dense deposits between the basement membrane and the epithelial cells, appearing as irregular spikes, which are best seen by silver stains as black in color .These spikes by time thicken to produce dome-like protrusions.
Immunofluorescence microscopy demonstrates the granular deposits to contain immunoglobulins and various amounts of complement.
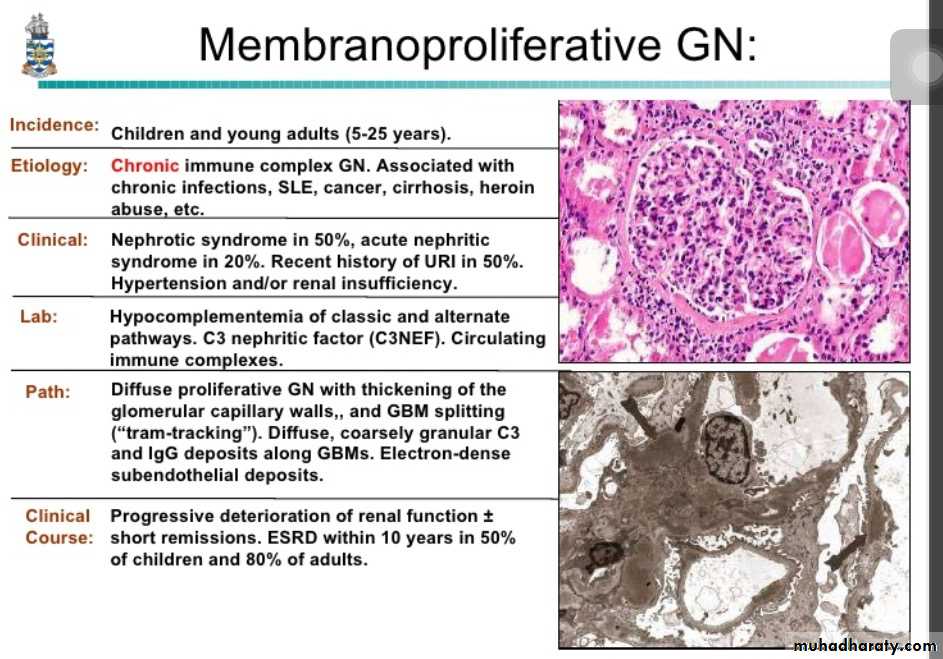
Membranoproliferative Glomerulonephropathy:
Sometimes referred as mesangioproliferative GN.MPGN is classified into; 1. Primary (idiopathic). 2. secondary, associated with;
Chronic immune complex disorders (SLE, hepatitis B and C infection, HIV infection).
Alfa1-antitrypsin deficiency.
Malignant tumors (leukemia, lymphoma).
Hereditary deficiency of complement regulatory proteins.
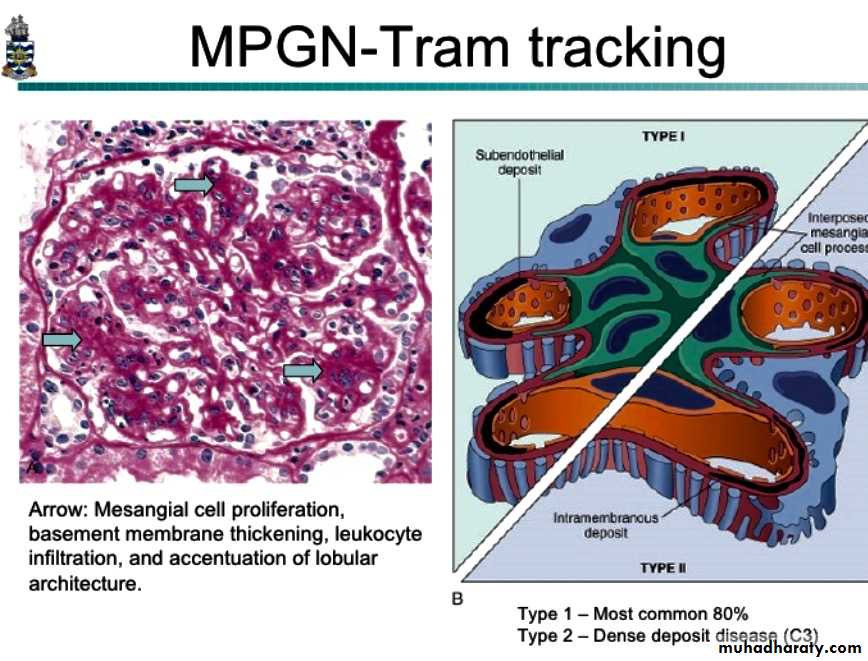
Types of MPGN
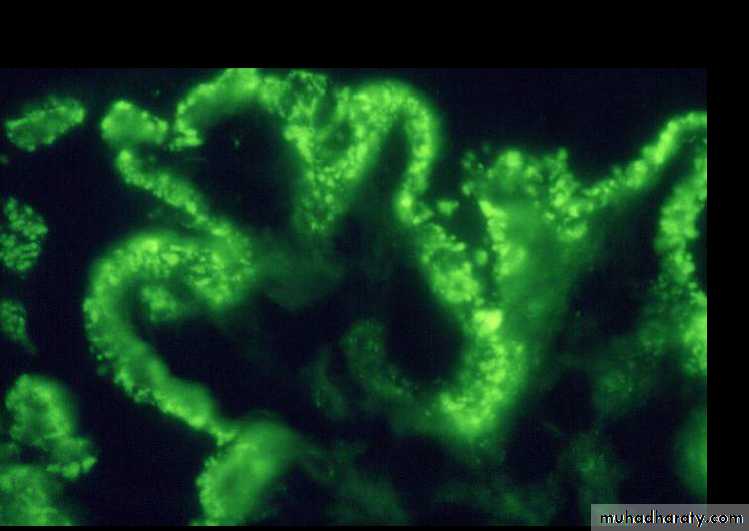
1.Type I (great majority of cases), is characterized by the presence of subendothelial electron-dense deposits. By the immunofuorescence, C3 is deposited in a granular pattern, and IgG and early complement components (C1q and C4) are often present, suggesting an immune complex pathogenesis.• Type II (dense-deposit disease), the GBM is transformed into an irregular, ribbon-like, extremely electron-dense structure, because of the deposition of dense material of unknown composition in the GBM proper. C3 is present in granular or linear pattern. IgG is usually absent, as are the early complement factors
Pathogenesis,
In most cases of type I MPGN, there is evidence of immune complexes in the glomerulus and activation of both classical and alternative complement pathways.Most patients with type II MPGN, have abnormalities that suggest activation of the alterative complement pathway.
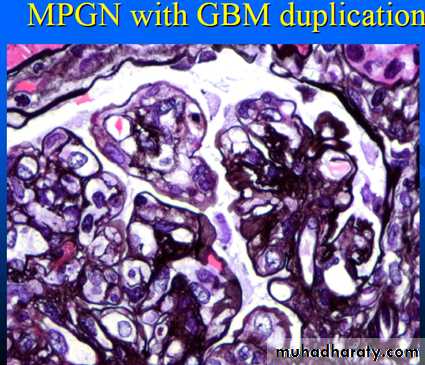
By light microscopy, the glomeruli are large due to hypercellularity secondary to proliferation of measngial cells, capillary endothelial cells and leukocytic infiltration.
The glomeruli have a lobular appearance
The GBM is clearly thickened. The glomerular capillary wall often shows a double contour or "tram-track" appearance.
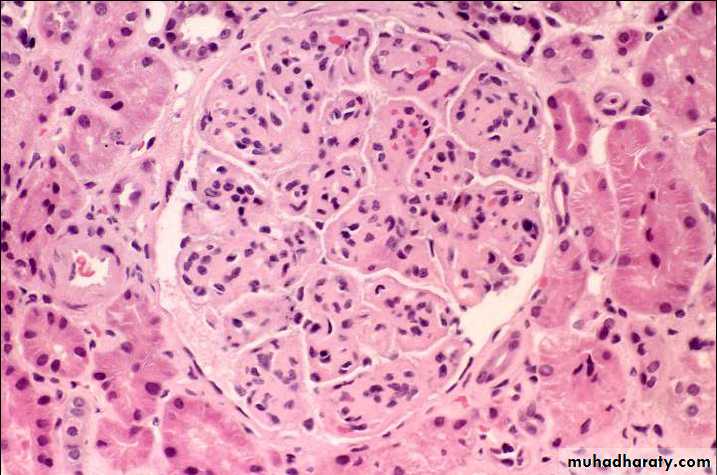
Focal Segmental Glomerulosclerosis
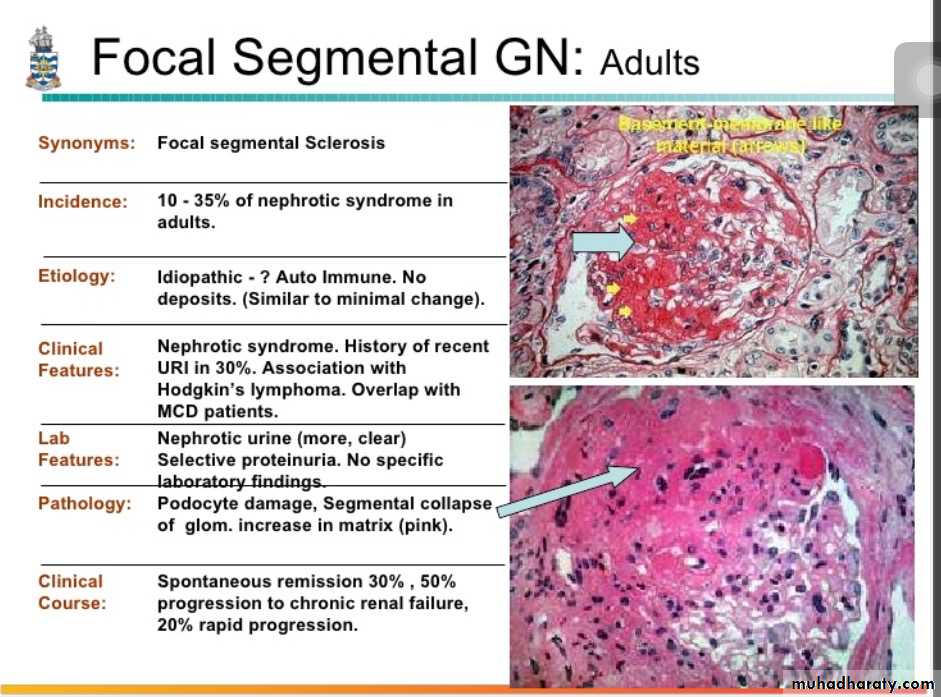
It occurs in the following settings;1. In association with other conditions (HIV infection, heroin addiction, sickle cell disease and massive obesity).
2. Secondary to glomerular disease (e.g. IgA nephropathy).
3. As a component of the adaptive response to loss of renal tissue (renal ablation).
4. Idiopathic (primary disease)..
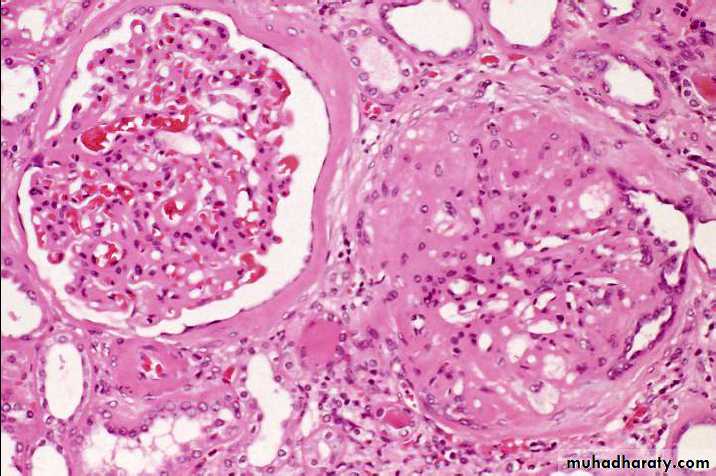
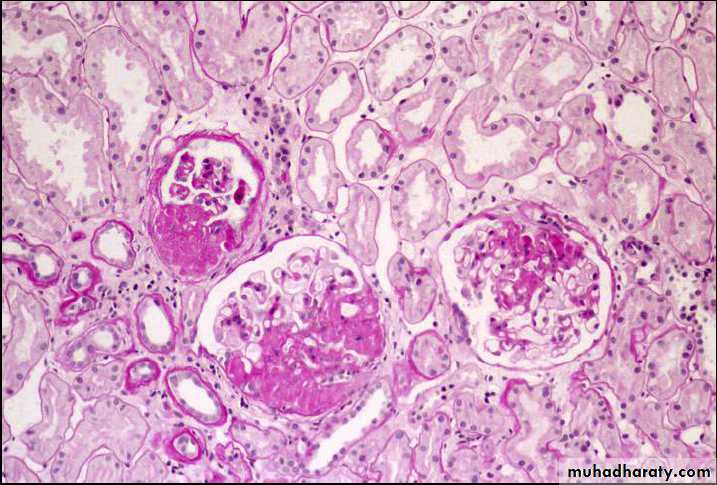
By light microscopy, the lesion may involve few glomeruli.
In the sclerotic segments, there is collapse of basement membranes, and increase in matrix.By electron microscopy, both sclerotic and non-sclerotic glomeruli show diffuse loss of foot processes of visceral epithelial cells and in addition, there may be focal detachment of the epithelial cells.
By Immunofluorescence microscopy, IgM and C3 may be present in the sclerotic areas and/or in the mesangium.
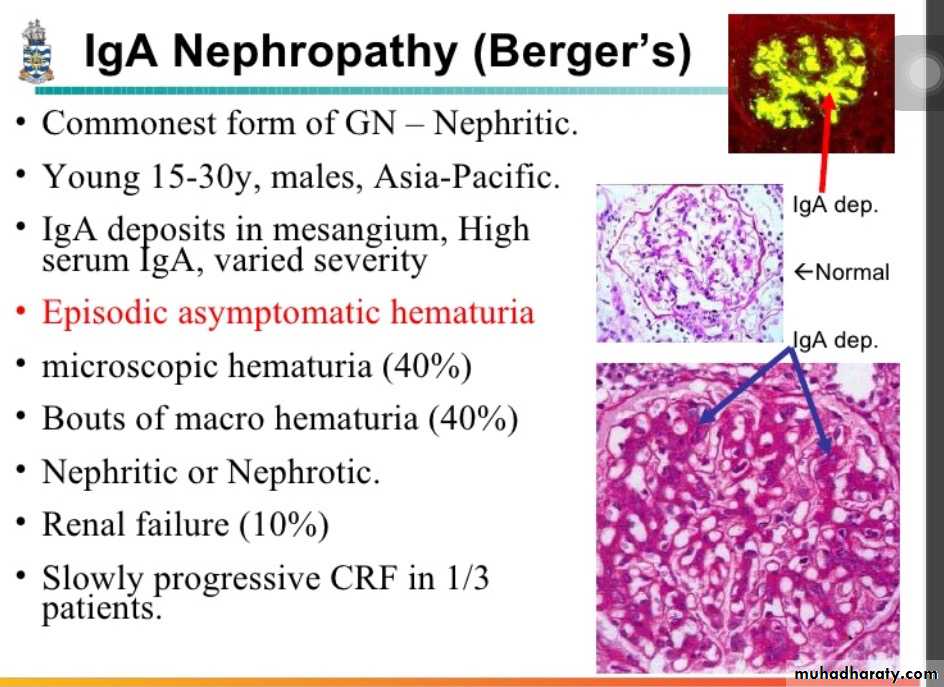
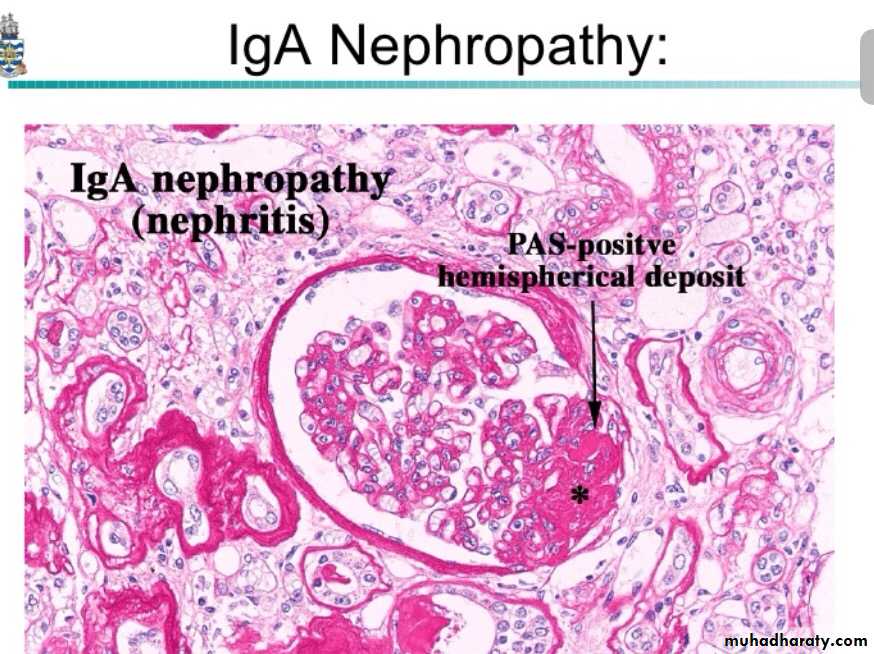
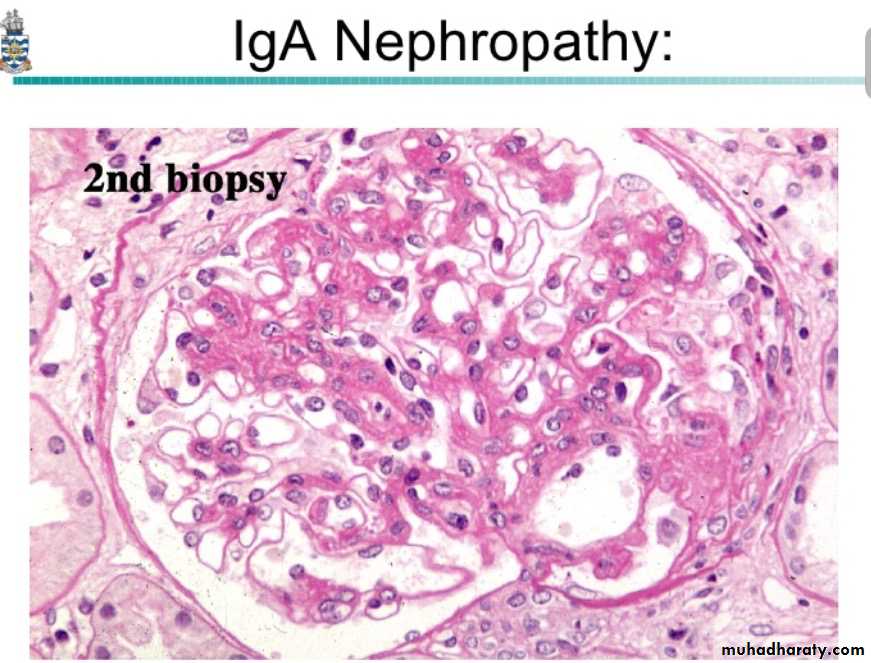
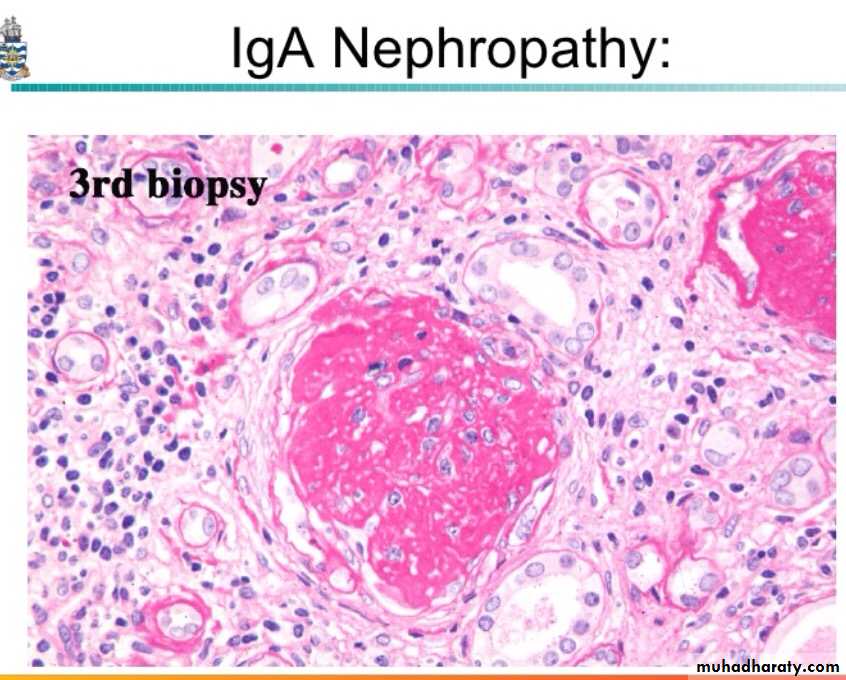
By light microscope, there is variable mesangial proliferation +/- sclerosis.
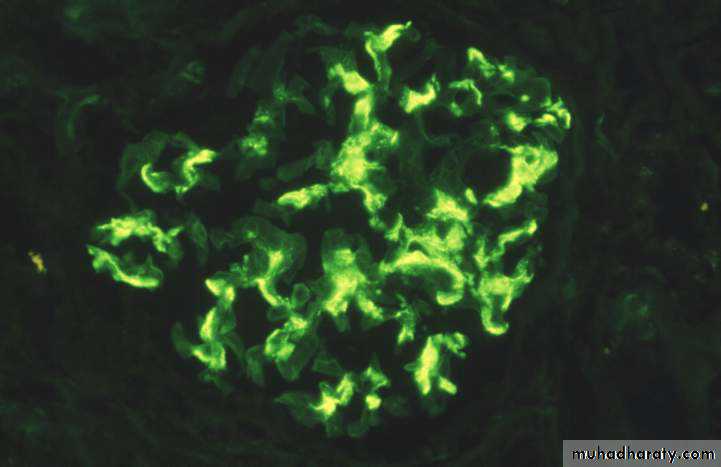
The characteristic picture is seen under fluorescent microscopy in form of mesangial deposition of IgA often with C3.
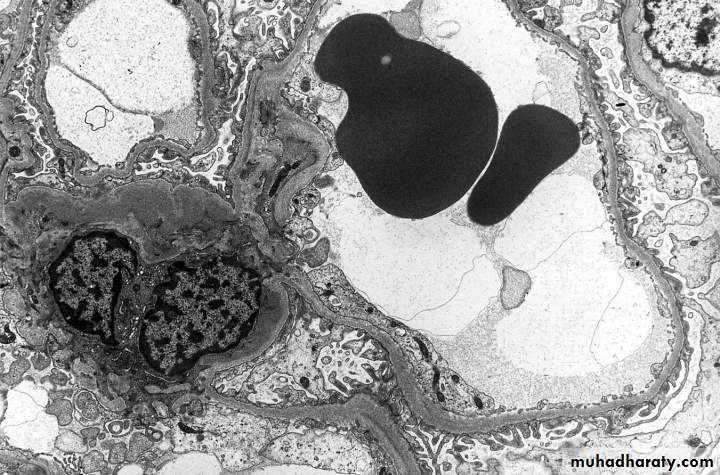
Electron microscopy confirms the presence of electron dense deposits in the mesangium.
Clinical courseThe disease affects people of any age, but older children and young adults are most commonly involved.
Many patients present with gross hematuria after an infection of respiratory tract, or GIT
Many patients maintain normal renal function for decades. Slow progression to chronic renal failure occurs in 15-40% of cases over a period of 20 years.Recurrence of IgA deposits in transplanted kidneys is frequent.
Chronic Glomerulonephropathy
Is best considered as a pool of end-stage glomerular disease.Morphology;
The kidneys are asymmetrically contracted and have diffusely granular cortical surfaces.
The cortex is thinned,
Light microscopically, there is hyaline obliteration of many glomeruli, arterial with arteriolar sclerosis, marked atrophy of associated tubules, and interstitial inflammation and fibrosis.
Clinical course
In most patients, chronic glomerulonephropathy develops insidiously and slowly progresses to renal insufficiency.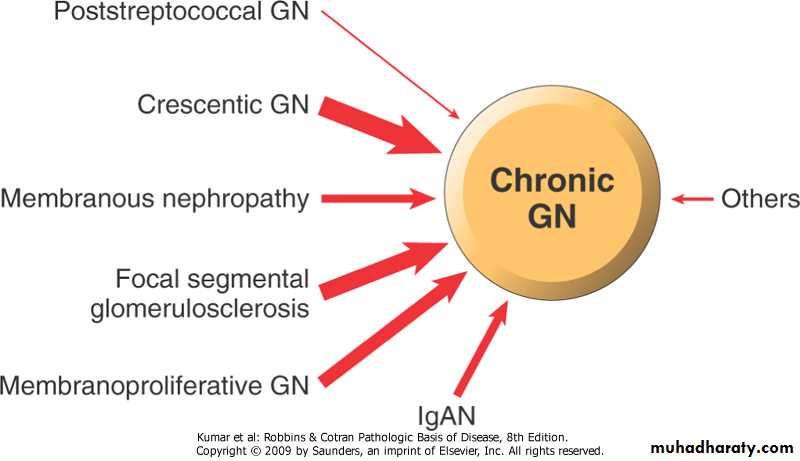
Chronic GN: Defined by the presence of > 2/3 sclerotic glomeruli