Rheumatology L-8

Patients who have thrombosis(arterial or venous) and/or recurrent miscarriages and who also have persistently positive blood tests for antiphospholipid antibodies(APAs) have the antiphospholipid syndrome(APS).
APS - no associated rheumatic disease (50%)= primary APS
APS - associated rheumatic diseasepresent in 10% of SLE patients=secondary APS
aPL - antiphospholipid antibodies (no symptoms)
CAPS - catastrophic APS ~ 238 worldwide reported casesAPAs can be detected by several tests:
*anticardiolipin test, which detects antibodies (IgG or IgM) that bind to negatively charged phospholipid, cardiolipin.*the lupus anticoagulant test which detect changes in the ability of the blood to clot in a test tube. This test act as a procoagulant factor inside the body. False positive tests for syphilis that found in patients with SLE are actually caused by APAs
*the anti-B2-glycoproten1 test, which detects antibodies that bind B2-glycoproten1, a molecule that interacts closely with phospholipids.
A persistently positive test( i:e. positive on at least two occasions, 6 weeks or more a part) in one or more of these assays is needed to diagnose APS.
However, some people who test positive for APAs will never get APS, i:e. not all APAs are harmful.
According to these facts, APS can be defined as a disorder that characterized by any or all of the fallowing three manifestations in the setting of positive antiphospholipid antibody tests:
1- recurrent arterial & or venous thrombosis
2- thrombocytopenia3- recurrent spontaneous abortions
ETIOLOGY:
APAs are directly pathogenic, but the exact mechanism of action is unknown.The presence of other co-factors for the development of APS may explain why subset of people can develop this disease than others . The most important co-factor is B2-glycoprotien.
b2-glycoprotein I
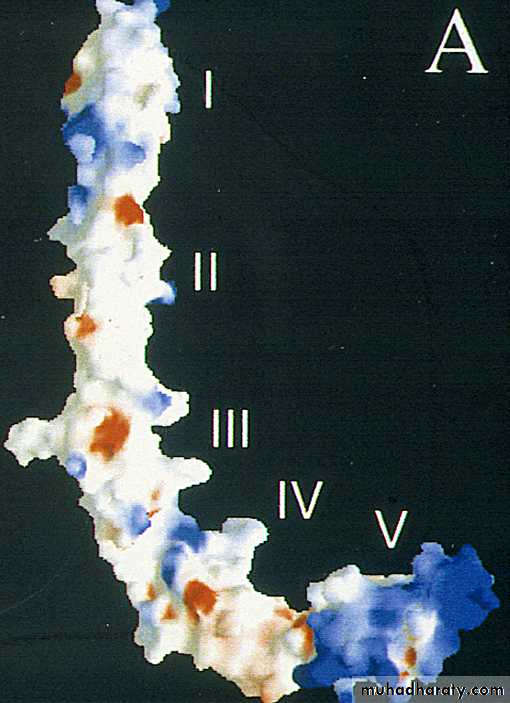
structure : 5 "sushi" domains
synthesis : liver
function : "in vitro" anticoagulantcongenital deficiency : asymptomatic
CLINICAL FEATURES:DEFENITIVE FEATURES
- arterial thrombosis - venous thrombosis -thrombocytopenia
-recurrent abortions
POSSIBLE FEATURES
- hemolytic anemia- livedo reticularis
- Leg ulcers
- chorea
- transverse myelitis
- vasculitis
- cardiac valvular malformations
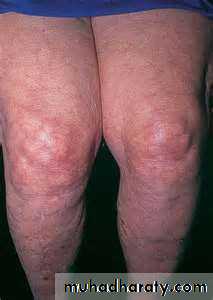
Antiphospholipid syndrome. Livedo reticularis
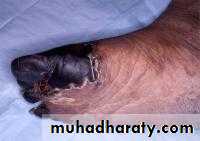
Catastrophic APS
describes patients who present with multiple thromboses, positive APAs, & often life threatening illness. Treatment requires cyclophosphamide& plasmapharesisDIAGNOSIS:
is made by clinical features & lab. evidence of APAs . the diagnosis should not be made unless at least one of the major criteria of APS is present.APAs are of two types: anticardiolipin & Lupus anticoagulant. Any patient with APS should be screened for SLE or other autoimmune diseases.
Lupus anticoagulant results in prolonged partial thrmboplastin time, but PTT is not a screening test for lupus anticoagulant.
TREATMENT:
NO cure from APS & NO single treatment is estimated & the treatment is directed into each manifestation.In patients with APS who have had one or more thrombosis, the recommended treatment to prevent further thrombosis is long term anticoagulation with warfarin.
Pregnant ladies with APS are given oral aspirin and subcutaneous heparin earlier in gestation to reduce the chance of abortion but pre-eclampsia and poor fetal growth remain common. Thrombocytopenia often does not need treatment, but it usually respond to steroids, IV Ig, danasol, splenectomy& immunosuppressive agents
.
SJOGREN'S SYNDROME
IS a chronic immune mediated inflammatory disorder of exocrine glands& other systemic features . The most common manifestations include inflammation & destruction of lacrimal & salivary glands leading to dry eyes(keratoconjunctivitis sicca or xerophalmia)& dry mouth(xerostomia).It associated with various autoantibodies& systemic features like interstitial lung diseases, vasculitis & lymphoma. Patients considered to have secondary Sjogren,s if they have another autoimmune disease such as SLE, RA, Scleroderma, primary biliary cirrhosis & to have primary Sjogren's if they have no underlying immune disease .
It is a disease of both males &females, although females are common patients. All races, ages ðnicities are affected.
DIAGNOSIS
diagnostic criteria1- subjective & objective evidence of keratoconjunctivitis sicca & xerostomia
2- presence of at least one of the fallowing 4 autoantibodies:
antinuclear antibodies, Rheumatoid factor, anti- Ro(SS-A)& anti-La(SS-B) antibodies
3- exclusion of any disease that may mimic Sjogren's syndrome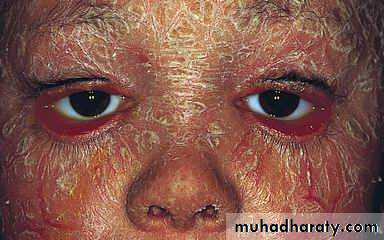
Schirmer Test
Normal :≥10mm/5min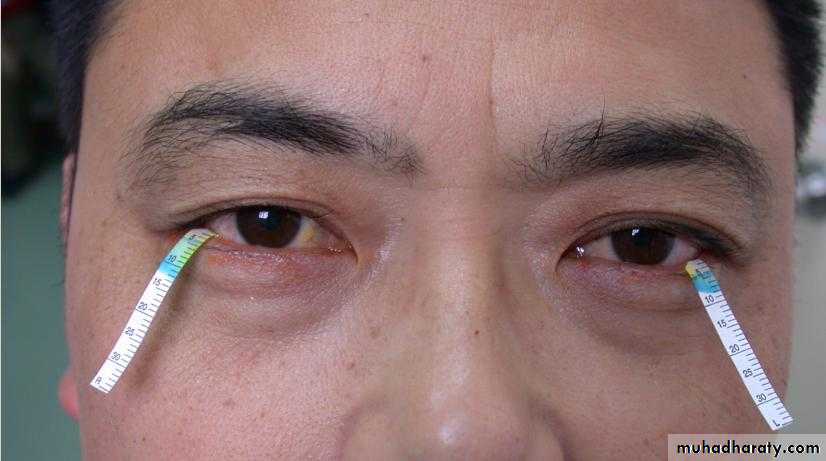
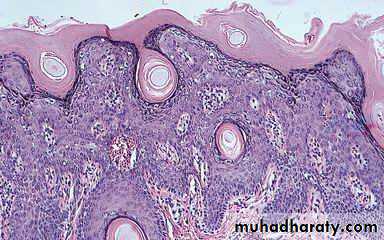
Biopsy from involved salivary glands may demonstrate the characteristic findings of focal lymphocytic infiltration of CD-4 T-cells which imply that cell mediated immunity is essential in the pathogenesis of Sjogren's syndrome.
MANAGEMENT
Generally speaking the management of systemic manifestations is the same as other autoimmune diseases & include steroids, antimalarials , & immunosuppressive agents. For xerostomia ; artificial saliva& avoidance of anticholinergic drugs. For xerophalmia we use artificial tears & eye glasses.Inflammatory myopathies
Rare heterogeneous group of acquired diseases characterized by inflammatory infiltrate of skeletal muscles. They are Potentially treatable.Bimodal age distribution in PM/DM
Between 10-15 years in children
Between 45-60 in adults
Inclusion body
More common after the age of 50
Female predominance
The etiology is unknown with variable genetic association. The commonest forms are poliomyelitis, dermatomyositis& inclusion body myositis.
Usually only skeletal muscles are affected. Sometimes, the myositis is focal( orbital myositis).
There is an increased incidence of malignancy in patients with dermatomyositis(3 fold) , polymyositis(30% increase). Malignancy may be apparent at the time of diagnosis or appears later.
The classification of Bohan et al(1977) is as fallows:
Group1: Primary idiopathic polymyositis.Group 2:primary idiopathic dermatomyositis.
Group 3:dermatomyositis(or polymyositis) associated with neoplasia.
• Group 4: childhood dermatomyositis (or polymyositis) associated with vasculitis.
• Group 5: polymyositis(or dermatomyositis) with associated collagen vascular disease.Polymyositis symmetrical proximal muscle weakness usually of the lower extremities first. The patient have difficulty in rising from a chair, climbing stair or lifting. Sometimes associated with muscle pain. The onset is between 40-60 years& is gradual, but can be insidious or explosive.
Fever, weight loss& fatigue are common. Respiratory & pharyngeal muscle involvement with respiratory failure& aspiration can occur & considered an emergency. Interstitial lung disease occurs in 30% & strongly associated with Jo-1 antibodies.
Dermatomyositis the muscle manifestations are similar to those of polymyositis but occur in combination with characteristic cutaneous manifestations:
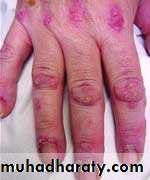
Gottron's papules are violaceous plaques or papules, scaly, erythematosus on the extensor surface of proximal & distal interphalangeal joints.
Dermatologic manifestations
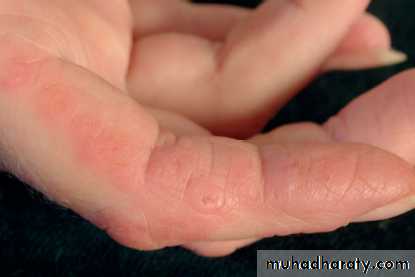
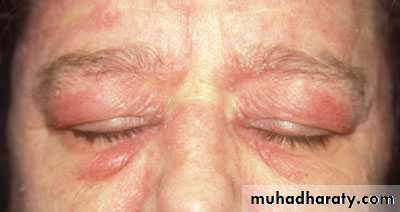
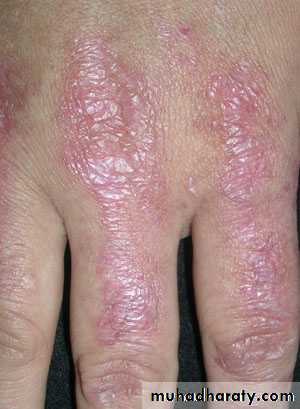
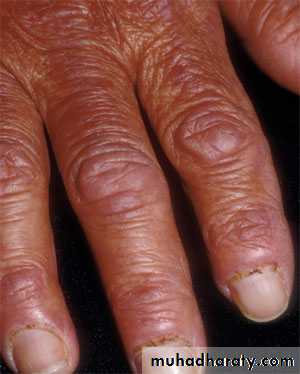
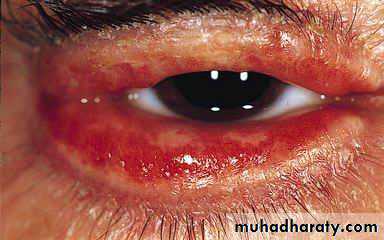
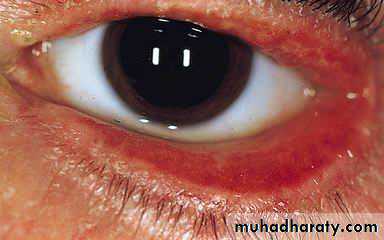
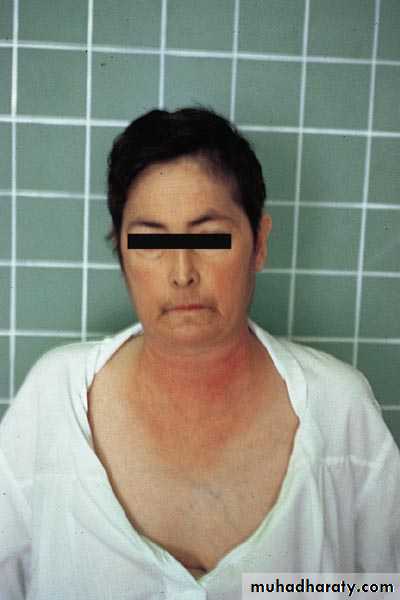
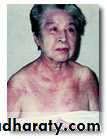
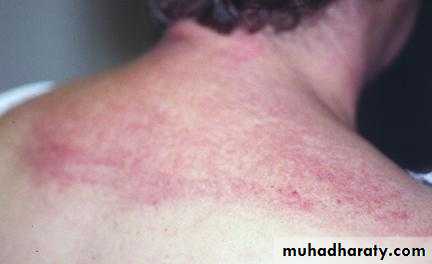
Heliotrope rash is a violaceous discoloration of eye lid with periorbital edema. similar rashes occur on the upper back, chest& shoulders('shawl' distribution). Fever ,weight loss& arthralgia also occur.
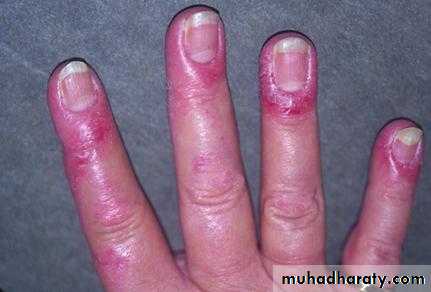
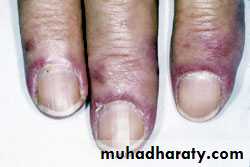
INVESTIGATIONS the diagnosis of IIM is suspected by typical history& physical examination. Muscle enzymes such as creatine phosphokinase(CPK) & aldolase are markedly elevated. The diagnosis is confirmed by muscle biopsy.
Ideally open biopsy is optimal, but fine needle muscle biopsy may be adequate.
Involved not atrophic muscle should be used. Commonly quadriceps or deltoid muscle is used.
TREATMENT corticosteroids are the gold standard in the treatment of inflammatory myopathies. 60 mg / day prednisolone is begun until CPK has returned to normal or muscle strength is greatly improved . Occasionally, extremely high dose IV corticosteroids are used for severely ill patients.
Gradual tapering of drug is attempted according to clinical response . One fourth to one third of patients require additional immunosuppressive drugs such as methotrexate or azathioprine because of steroid resistance, intolerable side effects or inability to taper steroids .
Cyclophosphamide & cyclosporine also used but they have toxic side effects.
Recently IV gamma globulin has proven effective in dermatomyositis . It works by blocking deposition of complement fragments in muscle capillaries , thereby prevents further injury. However, its long term efficacy is unknown & it is highly expensive.