
FIFTH STAGE
INTERNAL MEDICINE
DR.FADHIL – LECTURE 8
1
Anti-Phospholipid Antibody Syndrome
Patients who have thrombosis(arterial or venous) and/or recurrent miscarriages and
who also have persistently positive blood tests for antiphospholipid antibodies(APAs)
have the antiphospholipid syndrome(APS).
•
APS - no associated rheumatic disease (50%)= primary APS
•
APS - associated rheumatic disease present in 10% of SLE patients=secondary
APS
•
aPL - antiphospholipid antibodies (no symptoms)
•
CAPS - catastrophic APS ~ 238 worldwide reported cases
•
APAs can be detected by several tests:
•
*anticardiolipin test, which detects antibodies (IgG or IgM) that bind to
negatively charged phospholipid, cardiolipin.
•
*the lupus anticoagulant test which detect changes in the ability of the blood
to clot in a test tube. This test act as a procoagulant factor inside the body.
False positive tests for syphilis that found in patients with SLE are actually
caused by APAs
•
*the anti-B2-glycoproten1 test, which detects antibodies that bind B2-
glycoproten1, a molecule that interacts closely with phospholipids.
•
A persistently positive test( i:e. positive on at least two occasions, 6 weeks or
more a part) in one or more of these assays is needed to diagnose APS.
•
However, some people who test positive for APAs will never get APS, i:e. not all
APAs are harmful.
According to these facts, APS can be defined as a disorder that characterized by
any or all of the fallowing three manifestations in the setting of positive
antiphospholipid antibody tests:
1- recurrent arterial & or venous thrombosis
2- thrombocytopenia
3- recurrent spontaneous abortions

2
ETIOLOGY:
•
APAs are directly pathogenic, but the exact mechanism of action is unknown.
•
The presence of other co-factors for the development of APS may explain why
subset of people can develop this disease than others . The most important co-
factor is B2-glycoprotien.
B
2
-GLYCOPROTEIN I
•
structure : 5 "sushi" domains
•
synthesis : liver
•
function : "in vitro" anticoagulant
•
congenital deficiency : asymptomatic
CLINICAL FEATURES:
DEFENITIVE FEATURES
•
- arterial thrombosis - venous thrombosis -thrombocytopenia
•
-recurrent abortions
POSSIBLE FEATURES
•
- hemolytic anemia
•
- livedo reticularis
•
- Leg ulcers
•
- chorea
•
- transverse myelitis
•
- vasculitis
•
- cardiac valvular malformations
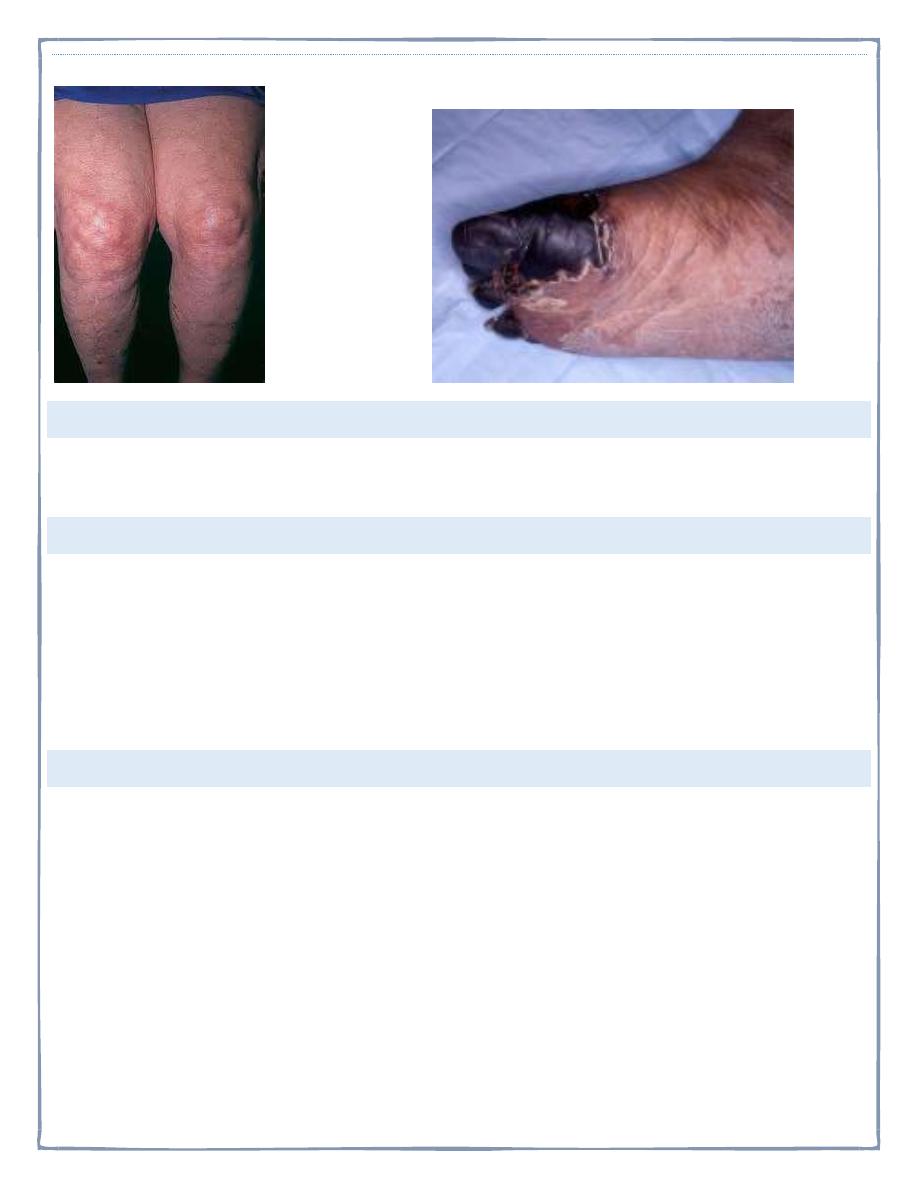
3
ANTIPHOSPHOLIPID SYNDROME. LIVEDO RETICULARIS:
CATASTROPHIC APS
Describes patients who present with multiple thromboses, positive APAs, & often life
threatening illness. Treatment requires cyclophosphamide& plasmapharesis
DIAGNOSIS:
• is made by clinical features & lab. evidence of APAs . the diagnosis should not
be made unless at least one of the major criteria of APS is present.
• APAs are of two types: anticardiolipin & Lupus anticoagulant. Any patient with
APS should be screened for SLE or other autoimmune diseases.
• Lupus anticoagulant results in prolonged partial thrmboplastin time, but PTT is
not a screening test for lupus anticoagulant.
TREATMENT:
• NO cure from APS & NO single treatment is estimated & the treatment is directed
into each manifestation.
• In patients with APS who have had one or more thrombosis, the recommended
treatment to prevent further thrombosis is long term anticoagulation with
warfarin.
• Pregnant ladies with APS are given oral aspirin and subcutaneous heparin earlier
in gestation to reduce the chance of abortion but pre-eclampsia and poor fetal
growth remain common. Thrombocytopenia often does not need treatment, but it
usually respond to steroids, IV Ig, danasol, splenectomy& immunosuppressive
agents
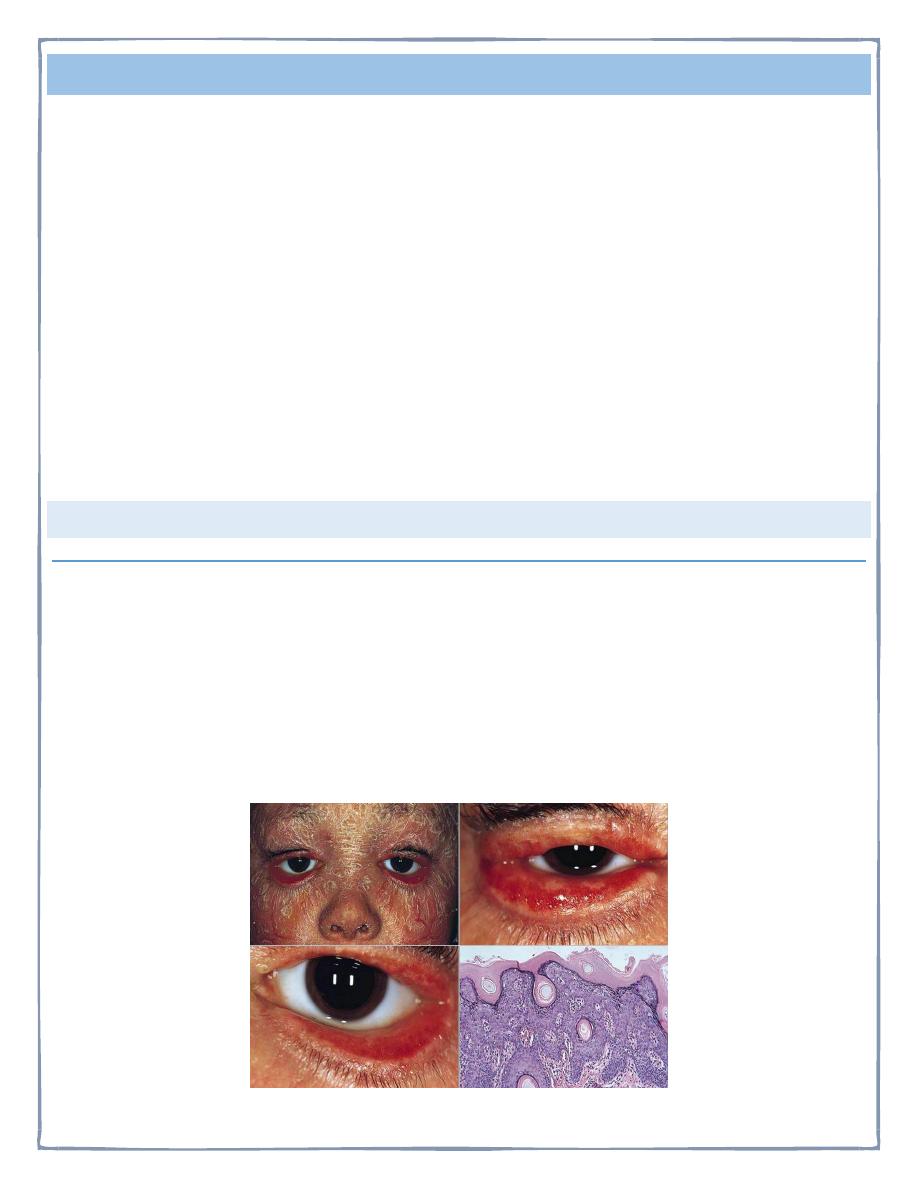
4
SJOGREN'S SYNDROME
•
IS a chronic immune mediated inflammatory disorder of exocrine glands&
other systemic features . The most common manifestations include
inflammation & destruction of lacrimal & salivary glands leading to dry
eyes(keratoconjunctivitis sicca or xerophalmia)& dry mouth(xerostomia).
•
It associated with various autoantibodies& systemic features like interstitial
lung diseases, vasculitis & lymphoma. Patients considered to have secondary
Sjogren,s if they have another autoimmune disease such as SLE, RA,
Scleroderma, primary biliary cirrhosis & to have primary Sjogren's if they have
no underlying immune disease .
•
It is a disease of both males &females, although females are common patients.
All races, ages ðnicities are affected.
DIAGNOSIS
DIAGNOSTIC CRITERIA
1- subjective & objective evidence of keratoconjunctivitis sicca & xerostomia
2- presence of at least one of the fallowing 4 autoantibodies:
• antinuclear antibodies, Rheumatoid factor, anti- Ro(SS-A)& anti-La(SS-B)
antibodies
3- exclusion of any disease that may mimic Sjogren's syndrome

5
SCHIRMER TEST:
Normal
:
≥10mm/5min
BIOPSY
from involved salivary glands may demonstrate the characteristic findings
of focal lymphocytic infiltration of CD-4 T-cells which imply that cell mediated
immunity is essential in the pathogenesis of Sjogren's syndrome.
MANAGEMENT:
•
Generally speaking the management of systemic manifestations is the same as
other autoimmune diseases & include steroids, antimalarials , &
immunosuppressive agents. For xerostomia ; artificial saliva& avoidance of
anticholinergic drugs. For xerophalmia we use artificial tears & eye glasses.
POLYMYOSITIS & DERMATOMYOSITIS
INFLAMMATORY MYOPATHIES
•
Rare heterogeneous group of acquired diseases characterized by inflammatory
infiltrate of skeletal muscles. They are Potentially treatable.
•
Bimodal age distribution in PM/DM
–
Between 10-15 years in children
–
Between 45-60 in adults
•
Inclusion body
–
More common after the age of 50
•
Female predominance

6
•
The etiology is unknown with variable genetic association. The commonest
forms are poliomyelitis, dermatomyositis& inclusion body myositis.
•
Usually only skeletal muscles are affected. Sometimes, the myositis is focal(
orbital myositis).
•
There is an increased incidence of malignancy in patients with
dermatomyositis(3 fold) , polymyositis(30% increase). Malignancy may be
apparent at the time of diagnosis or appears later.
THE CLASSIFICATION OF BOHAN ET AL(1977) IS AS FALLOWS:
Group1: Primary idiopathic polymyositis.
Group 2:primary idiopathic dermatomyositis.
Group 3:dermatomyositis(or polymyositis) associated with neoplasia.
Group 4: childhood dermatomyositis (or polymyositis) associated with vasculitis.
Group 5: polymyositis(or dermatomyositis) with associated collagen vascular
disease.
POLYMYOSITIS
symmetrical proximal muscle weakness usually of the lower extremities first. The
patient have difficulty in rising from a chair, climbing stair or lifting. Sometimes
associated with muscle pain. The onset is between 40-60 years& is gradual, but can
be insidious or explosive.
Fever, weight loss& fatigue are common. Respiratory & pharyngeal muscle
involvement with respiratory failure& aspiration can occur & considered an
emergency. Interstitial lung disease occurs in 30% & strongly associated with Jo-1
antibodies.
DERMATOMYOSITIS
the muscle manifestations are similar to those of polymyositis but occur in
combination with characteristic cutaneous manifestations:
Gottron's papules are violaceous plaques or papules, scaly, erythematosus on the
extensor surface of proximal & distal interphalangeal joints.
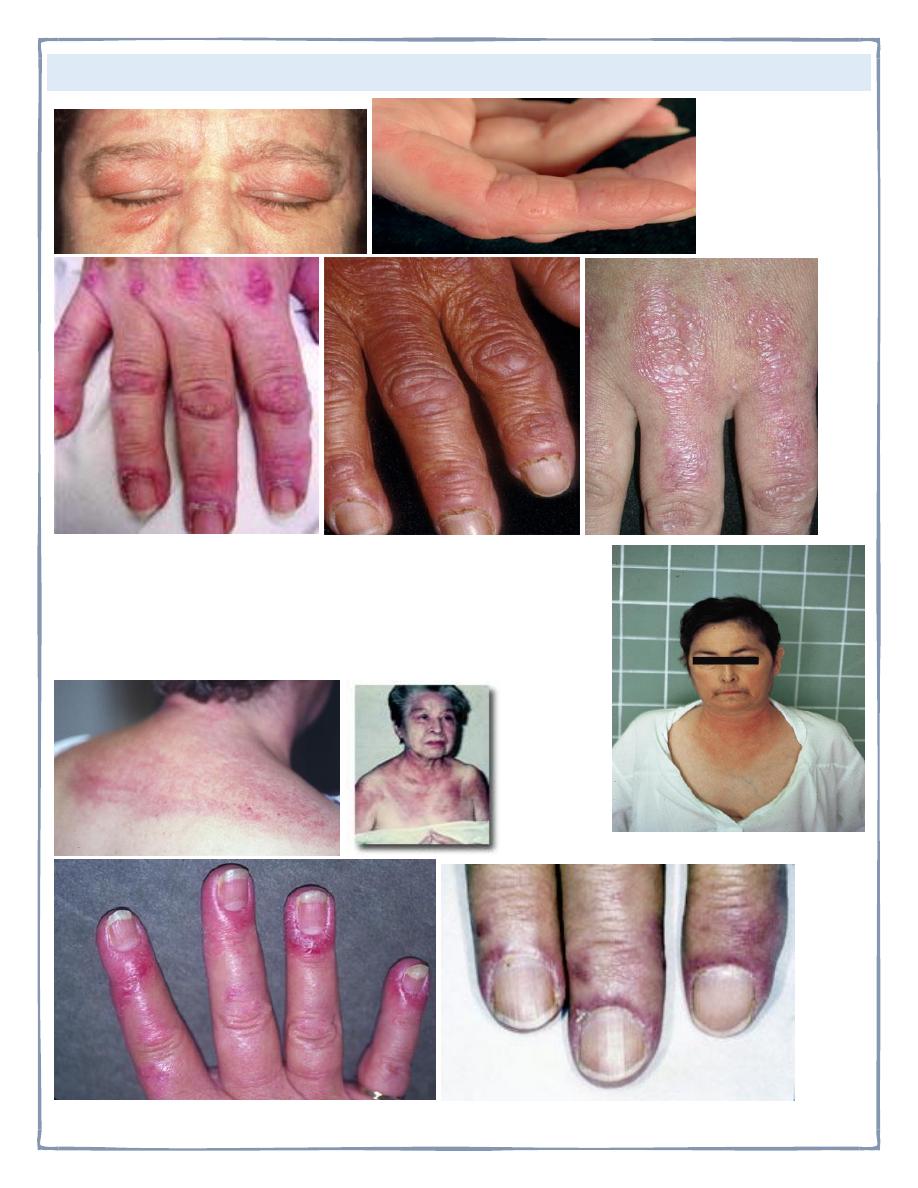
7
DERMATOLOGIC MANIFESTATIONS
Heliotrope rash is a violaceous discoloration of eye lid with
periorbital edema. similar rashes occur on the upper back,
chest& shoulders('shawl' distribution). Fever ,weight
loss& arthralgia also occur.

8
INVESTIGATIONS
• the diagnosis of IIM is suspected by typical history& physical examination.
Muscle enzymes such as creatine phosphokinase(CPK) & aldolase are markedly
elevated. The diagnosis is confirmed by muscle biopsy.
• Ideally open biopsy is optimal, but fine needle muscle biopsy may be adequate.
• Involved not atrophic muscle should be used. Commonly quadriceps or deltoid
muscle is used.
TREATMENT
•
corticosteroids are the gold standard in the treatment of inflammatory
myopathies. 60 mg / day prednisolone is begun until CPK has returned to
normal or muscle strength is greatly improved . Occasionally, extremely high
dose IV corticosteroids are used for severely ill patients.
•
Gradual tapering of drug is attempted according to clinical response . One
fourth to one third of patients require additional immunosuppressive drugs
such as methotrexate or azathioprine because of steroid resistance, intolerable
side effects or inability to taper steroids .
•
Cyclophosphamide & cyclosporine also used but they have toxic side effects.
•
Recently IV gamma globulin has proven effective in dermatomyositis . It works
by blocking deposition of complement fragments in muscle capillaries , thereby
prevents further injury. However, its long term efficacy is unknown & it is
highly expensive.
Thank you,,,