1
L6
Spinal Cord Disorder
D. Hazim
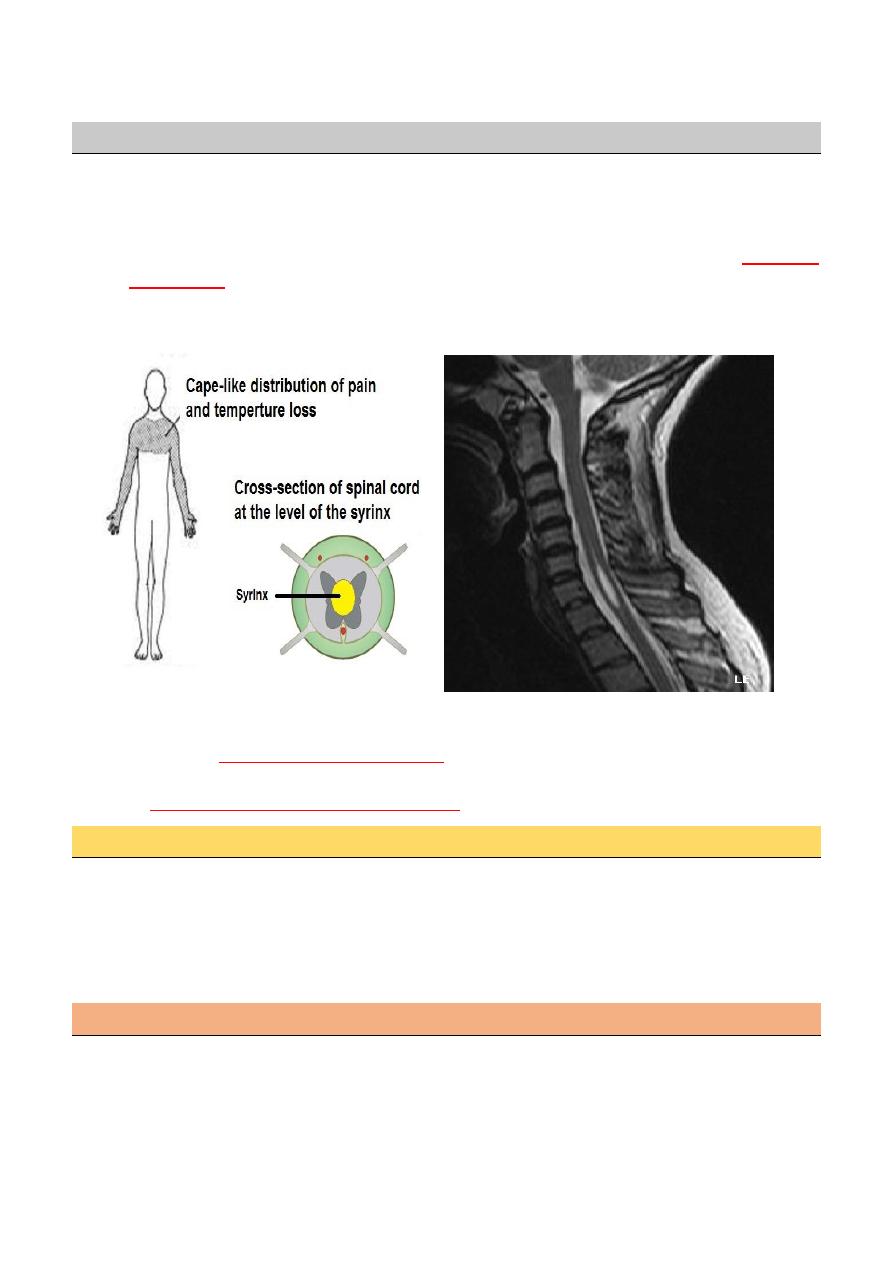
Syringomyelia
Cavitation of the spinal cord.
The symptoms and physical signs reflect a pathology that starts centrally and expands
outwards.
The cavity in syringomyelia affects crossing spinothalamic fibers producing a
half-cape
or cape loss
of pain and temperature sensation; posterior column signs are also found.
The patient complains of painless injuries, muscle wasting and weakness and more rarely
limb weakness.
Generally, there are two forms of syringomyelia:
Congenital
Arnold–Chiari malformation
Acquired the second major form of syringomyelia occurs as a complication
of
trauma, meningitis, hemorrhage, tumor
.
Clinical feature
There is amyotrophy(LMN) at the level of the cavity with tendon reflex loss.
In advanced stages Charcot joints develop.
Below the cavity there may be upper motor neuron symptoms and signs and disturbances
of sphincter function, which contrast with the lower motor neuron symptoms and signs at
the level of the syrinx.
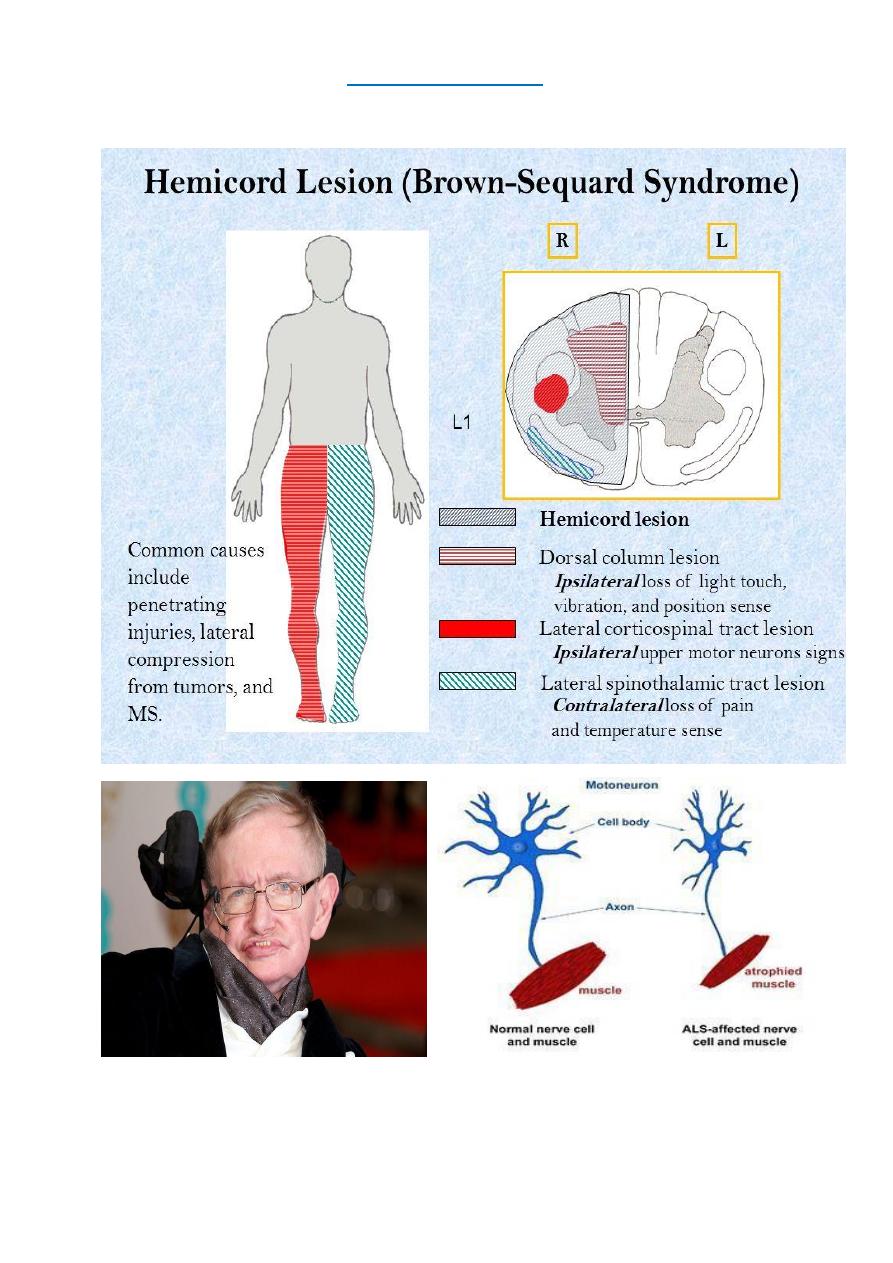
Brown- Sequard Syndrome
Brown- Séquard syndrome usually follows spinal cord hemisection as a result of penetrating
trauma can also occur with large disc herniation spinal epidural hematoma.
2
The classic description involves a
dissociated sensory loss
with contralateral loss of pain and
temperature but preserved ipsilateral light touch and posterior column function. In addition, there
is ipsilateral motor paralysis below the level of the lesion.
3
(MND) Motor neuron disease
Motor neuron disease e.g.(amyotrophic lateral sclerosis) is a progressive neuronal degenerative
disease that leads to severe disability and death begins usually above the ages of 50years .
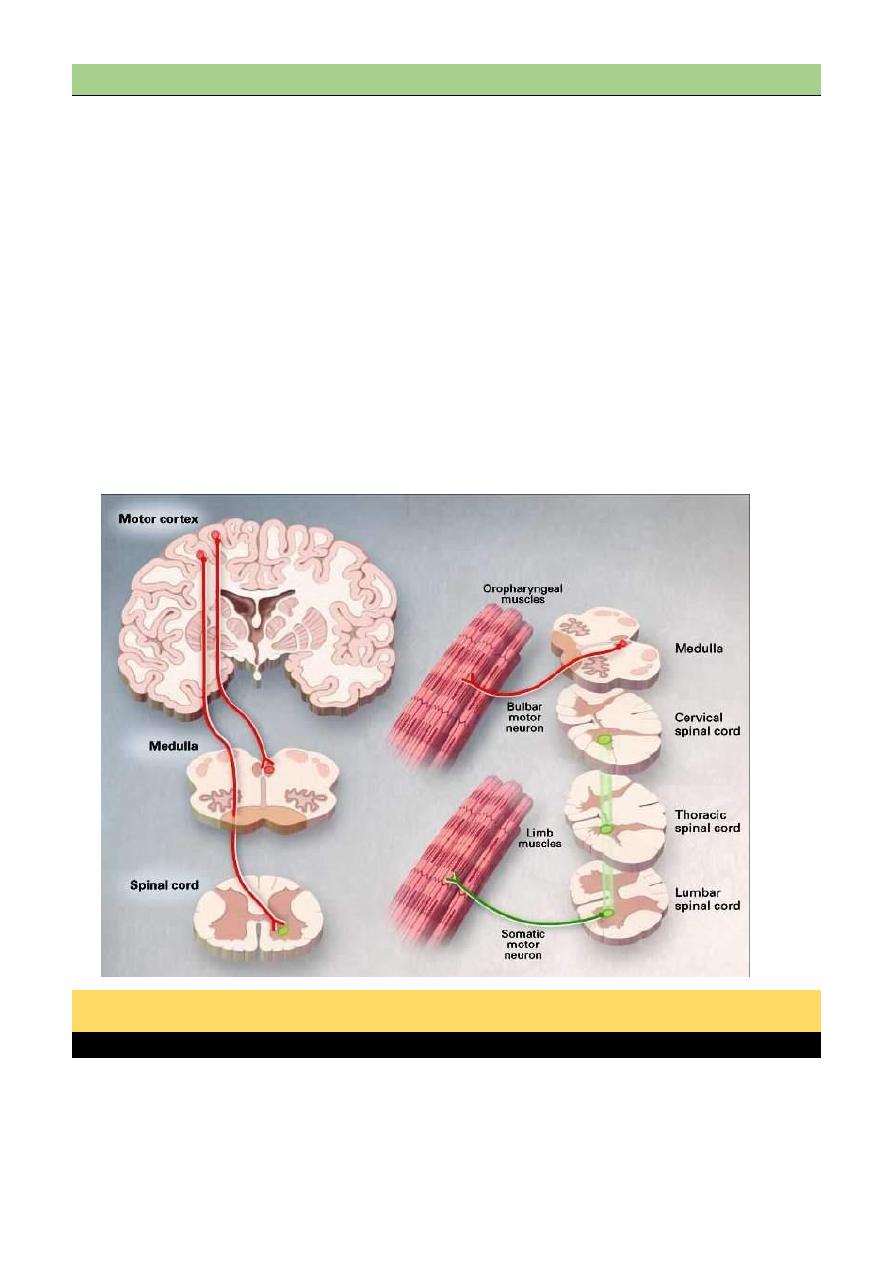
It is characterized by degeneration of anterior horn cells in the spinal cord, motor nuclei of the
lower cranial nerves in the brainstem, and corticospinal and corticobulbar pathways.
It is a disease in which certain nerve cells in the brain and spinal cord slowly die.
These nerve cells are called motor neurons, and they control the muscles that allow you to move
the parts of your body.
People who have MND gradually become more disabled, how quickly the disease gets worse is
different for everyone. Some people live with ALS for several years. But over time, ALS makes
it hard to walk, speak, eat, swallow, and breathe.
So there is features of combination of upper motor and lower motor type (characterized
clinically by wasting, weakness and fasciculation of the affected muscles with
hyperreflexia.
Patterns of Involvement of Motor Neuron Disease
Progressive muscular atrophy
Predominantly spinal motor neurons affected
Weakness and wasting of distal limb muscles at first
Fasciculation in muscles
Tendon reflexes may be absent

4
Progressive bulbar palsy
Early involvement of tongue, palate and pharyngeal muscles
Dysarthria/dysphagia
Wasting and fasciculation of tongue
May be pyramidal signs as well
Amyotrophic lateral sclerosis
Combination of distal and proximal muscle-wasting and weakness, fasciculation.
Spasticity, exaggerated reflexes, extensor plantars
Bulbar and pseudobulbar palsy follow eventually
Pyramidal tract features may predominate
Note
No Sensory Ivolvement
No Sphincter Dysfunction
No Ocular Muscles Affection
No Cerebellar Involvement
Investigation
The clinical features are highly suggestive
Electromyography helps to confirm the presence of fasciculation and denervation.
Spinal imaging and brain scanning may be necessary to exclude focal spinal or
cerebral disease.
CSF examination is usually normal.
Management
Riluzole, has recently been shown to have a small effect in prolonging life expectancy by about
two months.
Psychological and physical support.
Prognosis
The mean time from diagnosis to death is 1 year, with most patients dying within 3-5 years of
the onset of symptoms
Mubark A. Wilkins