Cystic Fibrosis (CF)
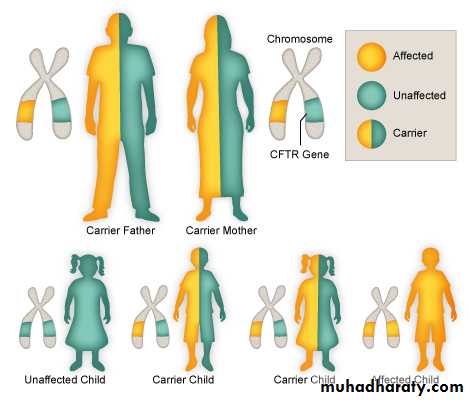
Cystic fibrosis (CF) is an inherited multisystem disorder.it is inherited as Autosomal Recessive.
the primary defect due to Dysfunction of the cystic fibrosis transmembrane conductance regulator protein (CFTR), leads to a wide and variable manifestations and complications.
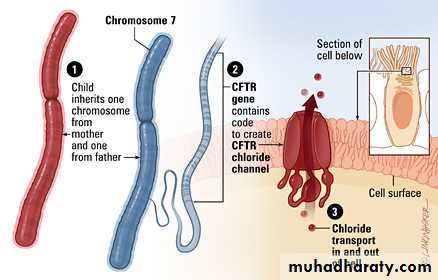
Gene encoding CFTR protein is located on long arm of chromosome 7 .
Large number of mutations responsible for CF but the most common is deletion of single phenylalanine residue at position 508 .pathogenesis
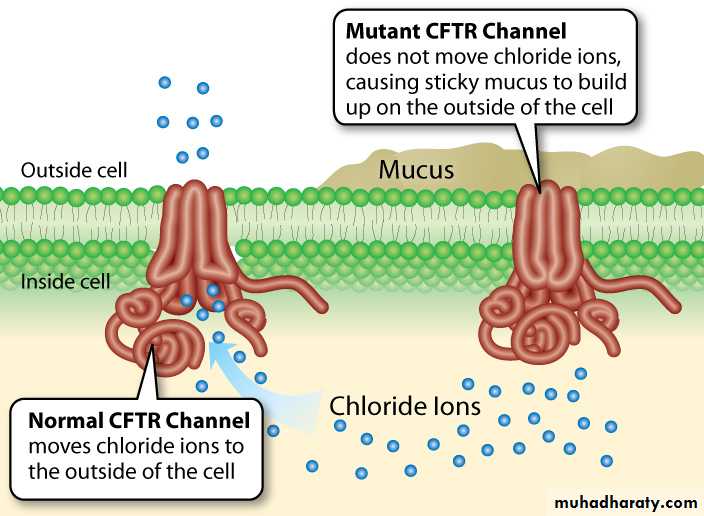
Dysfunction of CFTR inability to secrete Cl ion & excess absorption of Na across Resp.epithelial cell membrane movement of water from airway lumen to inside the cell insufficient water to hydrate secretions viscous secretions hard to clear by mucocilliary mechanism.
The result is that these secretions are retained and obstruct airways, starting with those of the smallest caliber, the bronchioles.
similar pathophysiologic events take place in the pancreatic and biliary ducts (and in the vas deferens), leading to thickening
of secretions and obstruction.
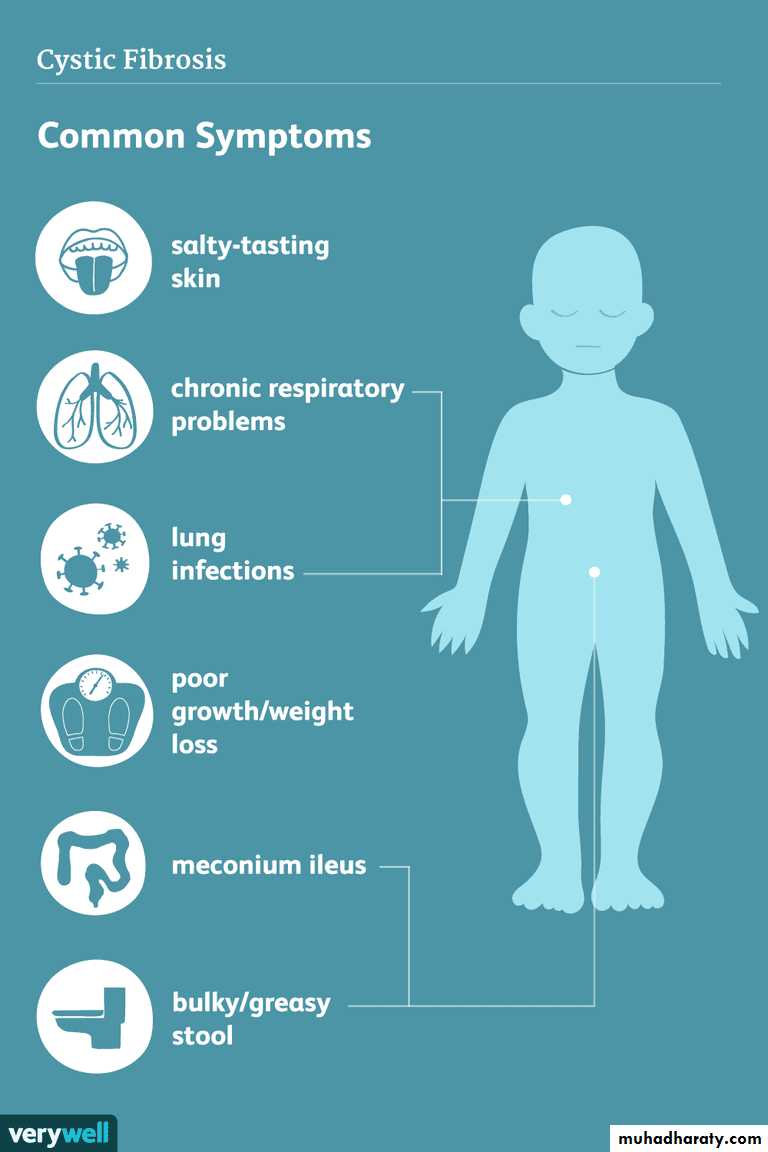
Because the function of sweat gland duct cells is to absorb rather than secrete chloride, salt is not retrieved from the isotonic primary sweat as it is transported
to the skin surface; chloride and sodium levels are consequently elevated.
CLINICAL MANIFESTATIONS
Resp. tract:Chronic productive cough of purulent mucous mainly at morning.
Recurrent sinupulmonary infections
Clubbing
Failure to thrive
Nasal polyps(˃ 5 years)
Atelectasis, corpulmonale, hemoptysis(beyond 1st decade)
Intestinal Tract:
Meconium ileus (neonate)
Distal intestinal obstruction syndrome(older child)
Exocrine pancreatic insuffeciency(steatorhea,FTT)
Fat soluble vitamins defeciency
Intususeption, rectal prolapse
Billiary Tract:
Neonatal jaundiceBilliary cirrosis and liver dysfunction.
Recurrent pancreatitis and hyperglycemia ( usualy in 2nd decade)
Genitourinary Tract:Azoospermia( in 95% of males),sexual function intact.
Thick cervical secretions in females, deminished fertility rate, pregnancy is well tolerated in women with normal lung function but can result in progression of lung disease & glucose intolerance.
Excessive loss of salt in sweat lead to salt depletion, hypochloremic alkalosis and hyponatremia especialy in warm climate and during episodes of gastroentritis.
Diagnosis
Diagnostic criteria for CFPresence of typical clinical features (respiratory, gastrointestinal,
or genitourinary) or A history of CF in a sibling or A positive newborn screening test
Plus
Two elevated sweat chloride concentrations obtained on separate days or Identification of two CF mutations or An abnormal nasal potential difference measurement
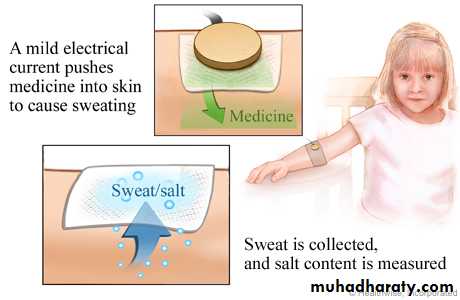
• 1)Sweat test:
• Sweat chloride ˃ 60 meq/l is diagnostic• 2) DNA analysis
• 3) increased potential differences across nasal epithelium
• 4) 72 hr fecal fat measurement & fecal elastase activity in fresh stool specimen (pancreatic exocrine dysfunction)
5) yearly monitoring with a modified 2 hr oral glucose tolerance test after 10 yr of age.
6) CXR , CT scan: bronchiectasis, patchy atelectasis, cyst formation.7) Pulmonary function test: started at 5 years of age.
8) Microbiologic studies: (H.influenzae, staph.aureus, pseudomonas, B.cepacia) commonly isolated from respiratory secretions. In addition to non tuberculous mycobacteria and aspergillus fumigatus( which cause allergic bronchopulmonary aspergillosis)
Newborn Screening
Newborn diagnoses can prevent early nutritional deficiencies and improve long-term growth and may improve cognitive function.Good nutritional status is associated with better lung function at 6 yr of age.
screening involve combination of immunoreactive trypsinogen (IRT) results and limited DNA testing on blood spots.
babies with an elevated IRT and a single detected mutation are considered a positive screen, and should be followed by a confirmatory sweat analysis.
CFTR -related metabolicsyndrome (CRMS)
infants with a positive newborn screen for CF who have a nondiagnostic sweat chloride (30-59 mmol/L)These infants should be followed closely through the 1st yr and then annually to evaluate them for the development of CF symptoms.
Treatment
Pulmonary Therapy:The goal is to release secretions and control infections:Chest physiotherapy
Encourage forceful cough and deep expiration
Antibiotics(oral, parentral, inhaled) for lung infections
Nebulized mucolytics eg N-acetylcystein,hypertonic saline, human recombant Dnase
Inhaled bronchodilators and steroids for reactive airway disease encountered in some patients with CF
Nutritional therapy:
Pancreatic Enzyme Replacement should not exceed 2,500 lipase units/kg/meal in most circumstancesinfants need 2,000-4,000 lipase units per feeding
Encourage high colories diet or formula
Nocturnal NGT feeding if still no wt gain
Supply fat soluble vitamins(especialy D3 1000 u/kg/week)
free access to water and salts especially during warm climates and during episodes of acute gastroenteritis
GH therapy in selected patients.
Ivacaftor (CFTR potentiator) approved for patients older than 2 yr of age with class III and class IV mutations.
prognosis
Median survival 40 years.male more favourable outcome than females.
Resp. complications is major determinant of mortality.