1
The liver and Gall bladder
Prof.Dr.Maha Shakir LEC1
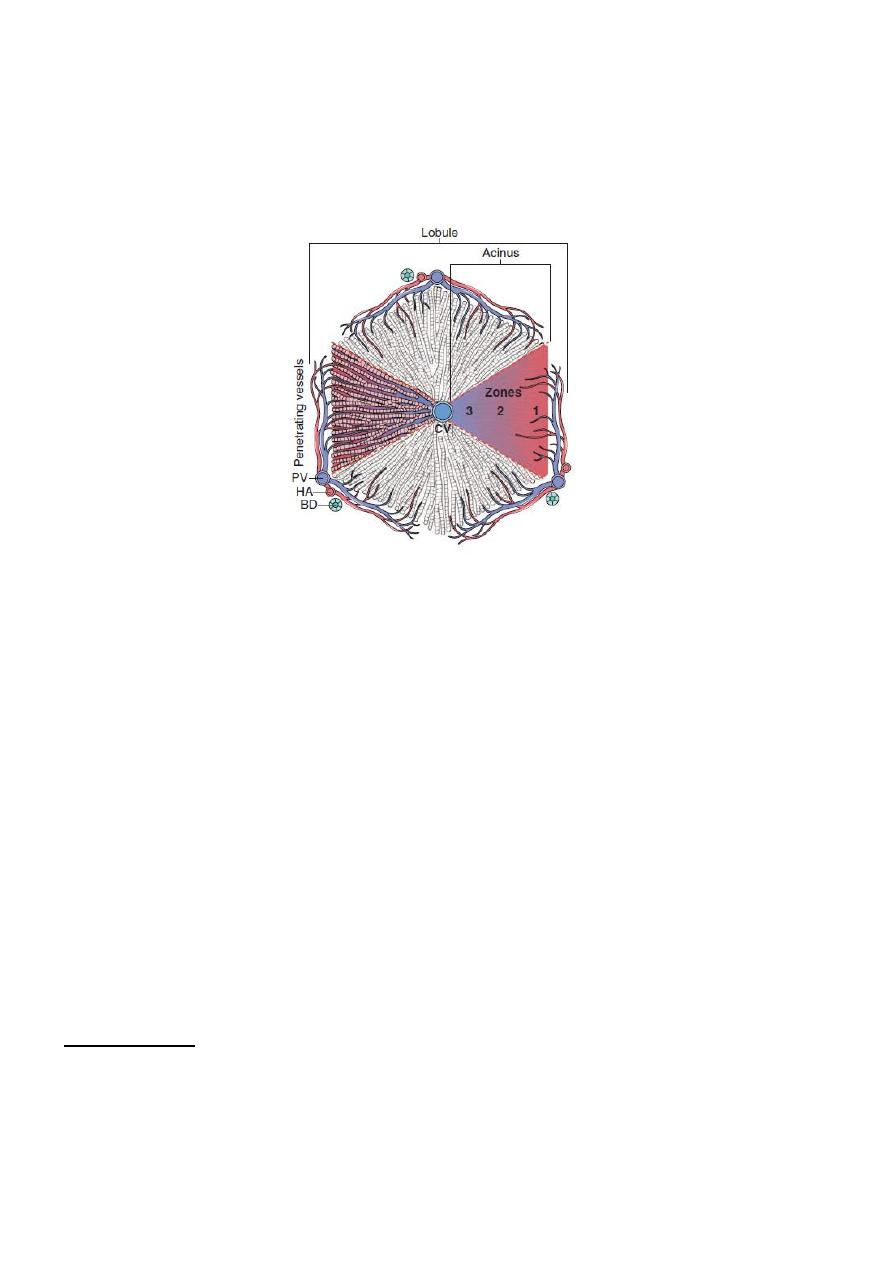
Fig. Models of liver anatomy. In the lobular model, the terminal hepatic vein is at the center of a
“lobule,” while the portal tracts are at the periphery
.
Pathologists often refer to the regions of the
parenchyma as “periportal and centrilobular.” In the acinar model, on the basis of blood flow, three
zones can be defined, zone 1 being the closest to the blood supply and zone 3 being the farthest. BD, Bile
duct; CV, central hepatic vein; HA, hepatic artery.PV, portal tracts.
Liver Failure
The most severe clinical consequence of liver disease is liver failure. It primarily
occurs in three clinical scenarios:acute, chronic, and acute-on-chronic liver failure.
Acute Liver Failure
Acute liver failure is defined as a liver disease that produces hepatic encephalopathy
within 6 months of the initial diagnosis. The condition is known as fulminant liver
failure when the encephalopathy develops within 2 weeks of the onset of jaundice,
and as subfulminant liver failure›when the encephalopathy develops within 3 months.
Hepatitis
Hepatitis
:-mean any
inflammatory lesion of the liver. This term is not used for
local lesions such as an abscess but only when there is diffuse involvement of the
liver.
There are many etiological factors that causing hepatitis such as alcoholic hepatitis,
drug induce hepatitis and viral hepatitis.
Viral hepatitis:- mean infection of the hepatocytes by virus that produces necrosis
and inflammation of the liver and presented with jaundice. It caused mainly by
hepatotropic viruses( which are hepatitis virus A,B,C,D,E,G).
Hepatitis viruses C &B are the most common cause of chronic hepatitis, liver
cirrhosis and hepatocellular carcinoma.

2
Clinicopathologic Syndromes of Viral Hepatitis
infection with hepatitis viruses produces a wide range of outcomes. Acute infection
by each of the hepatotropic viruses may be symptomatic or asymptomatic.
HAV and HEV do not cause chronic hepatitis, and only a small number of HBV-
infected adults develop chronic hepatitis. In contrast, HCV is notorious for producing
chronic infections.
Fulminant hepatitis is unusual and is seen primarily with HAV, HBV, or HDV
infections.
Although HBV and HCV are responsible for most cases of chronic hepatitis, there are
many other causes of similar clinicopathologic presentations, including autoimmune
hepatitis and drug- and toxin-induced hepatitis.
Acute viral hepatitis:-
Whichever virus is involved, acute disease follows a similar
course, consisting of:
(1) an incubation period of variable length
(2) a symptomatic preicteric phase;
(3) a symptomatic icteric phase;
(4) convalescence.
Peak infectivity occurs during the last asymptomatic days of the incubation period
and the early days of acute symptoms.
Pathological Features ot acute hepatitis
:-
Grossly, normal or slightly mottled. At the other end of the spectrum,massive hepatic
necrosis may produce a greatly shrunken liver.
Microscopically,
1. mononuclear cells predominate in all phases of viral hepatitis.
Portal inflammation in acute hepatitis is minimal or absent.
2. hepatocyte injury may result in necrosis or apoptosis. In severe acute hepatitis,
confluent necrosis of hepatocytes is
seen around central veins.
3. With increasing severity, there is central portal bridging necrosis, followed by
parenchymal collapse.
4. In its most severe form, massive hepatic necrosis and fulminant liver failure
ensue.
Chronic Hepatitis:-
The defining histologic feature of chronic viral hepatitis is mononuclear portal
infiltration.
It may be mild to severe and variable from one portal tract to the other.
There is lobular hepatitis.
The hallmark of progressive chronic liver damage is scarring.
At first, only portal tracts exhibit fibrosis, but in some patients, with time, fibrous
septa—bands of dense scar—will extend between portal tracts. In the most severe
cases, continued scarring and nodule formation leads to the development of cirrhosis.
3
Certain histologic features point to specific viral etiologies in chronic hepatitis. In
chronic hepatitis B, “ground-glass” hepatocytes (cells with endoplasmic reticulum
swollen by HBsAg) are a diagnostic hallmark, and the presence of viral antigen in
these cells can be confirmed by immunostaining.
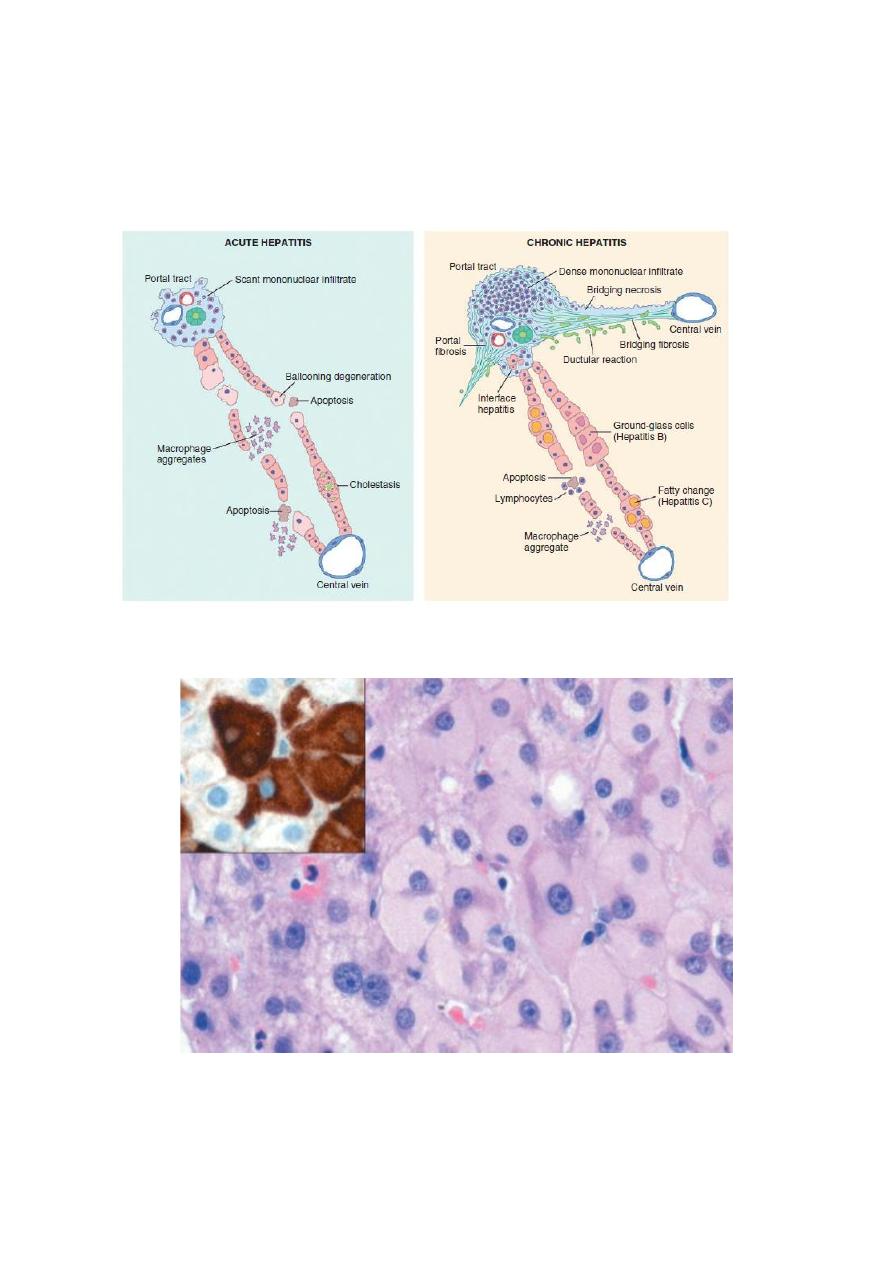
Fig. Morphologic features of acute and chronic hepatitis. There is very little portal mononuclear infiltration in acute
hepatitis (or sometimes none at all), while in chronic hepatitis portal infiltrates are dense and prominent—the defining
feature of chronic hepatitis. Bridging necrosis and fibrosis are shown only for chronic hepatitis, but bridging necrosis
may also occur in more severe acute hepatitis.
Fig. Ground-glass hepatocytes in chronic hepatitis B, caused by accumulation of hepatitis B surface antigen.
Hematoxylin-eosin staining shows the presence of abundant, finely granular pink cytoplasmic inclusions;
immunostaining (inset) with a specific antibody confirms the presence of surface antigen (brown).
Notes for VIRAL HEPATITIS
• (hepatitis A and E) never cause chronic hepatitis, only AcutE hepatitis.

4
Only the consonants (hepatitis B, C, D) have the potential to cause chronic disease .
• Hepatitis C is the single virus that is more often chronic than not (almost never
detected acutely; 85% or more of patients develop chronic hepatitis, 20% of whom
will develop cirrhosis).
• Hepatitis D, the delta agent, is a defective virus, requiring hepatitis B coinfection for
its own capacity to infect and replicate.
• The inflammatory cells in both acute and chronic viral hepatitis are mainly T cells.
• Patients with long-standing HBV or HCV infections are at increased risk for
development of HCC.
ALCOHOLIC AND NONALCOHOLIC FATTY LIVER
DISEASE
Alcohol is a well-known cause of fatty liver disease in adults and can manifest
histologically as steatosis, steatohepatitis, and cirrhosis. In recent years, it has become
evident that another entity, so-called “nonalcoholic fatty liver disease (NAFLD),” can
mimic the entire spectrum of hepatic changes associated with alcohol abuse. Since
the morphologic changes of alcoholic and NAFLD are indistinguishable, they are
discussed together
Three types of liver alterations are observed in fatty liver disease:
steatosis (fatty change), hepatitis (alcoholic or steatohepatitis), and fibrosis.
1. steatosis (fatty change):- accumulation of fat in hepatocytes (steatosis). The
pathogenesis of fatty liver is clearly depend on the intake of ethanol because it is
fully and rapidly reversible when the alcohol consumption is stopped.
Morphological Features:-
Grossly, fatty livers with widespread steatosis are large (weighing 4–6 kg or more),
soft, yellow, and greasy.
Microscopically: Hepatocellular fat accumulation typically begins in centrilobular
hepatocytes. The lipid droplets range from small (microvesicular) to large
(macrovesicular), the largest filling and expanding the cell and displacing the
nucleus.
As steatosis becomes more extensive, the lipid accumulation spreads outward from
the central vein to hepatocytes in the midlobule and then the periportal regions.
5
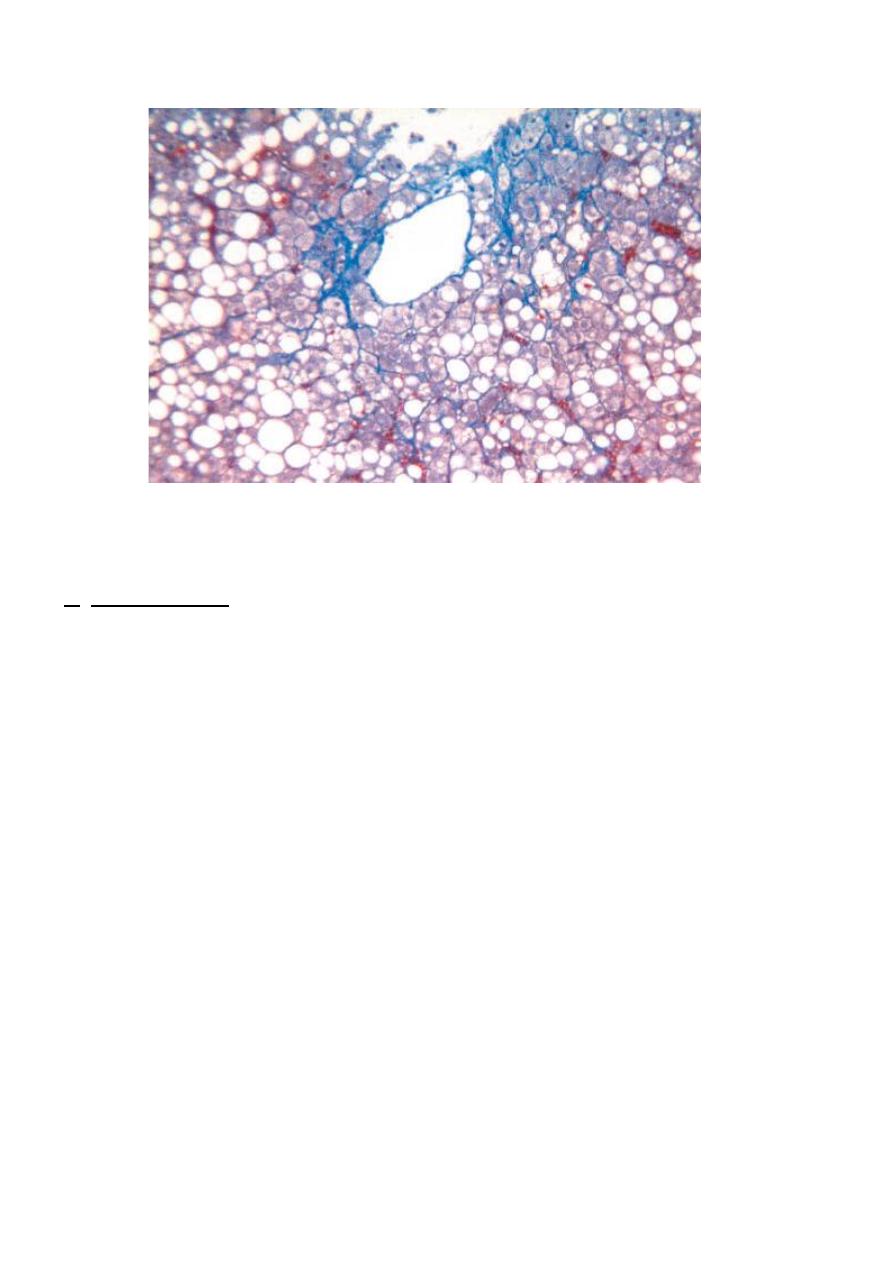
Fig. Fatty liver disease associated with chronic alcohol use. A mix of small and large fat droplets (seen as
clear vacuoles) is most prominent around the central vein and extends outward to the portal tracts. Some
fibrosis (stained blue) is present in a characteristic perisinusoidal
(Masson trichrome stain).
2. Steatohepatitis. These changes typically are more pronounced with alcohol use
than in NAFLD, but can be seen in either:
• Hepatocyte ballooning. Single or scattered foci of cells undergo swelling and
necrosis, these features are most prominent in the centrilobular regions
• Mallory-Denk bodies. These consist of tangled skeins of intermediate filaments
(including keratins 8 and 18) and are visible as eosinophilic cytoplasmic inclusions in
degenerating hepatocytes .
• Neutrophil infiltration. Predominantly neutrophilic infiltration may permeate the
lobule and accumulate around degenerating hepatocytes, particularly those containing
Mallory-Denk bodies. Lymphocytes and macrophages also may be seen in portal
tracts or parenchyma.
6
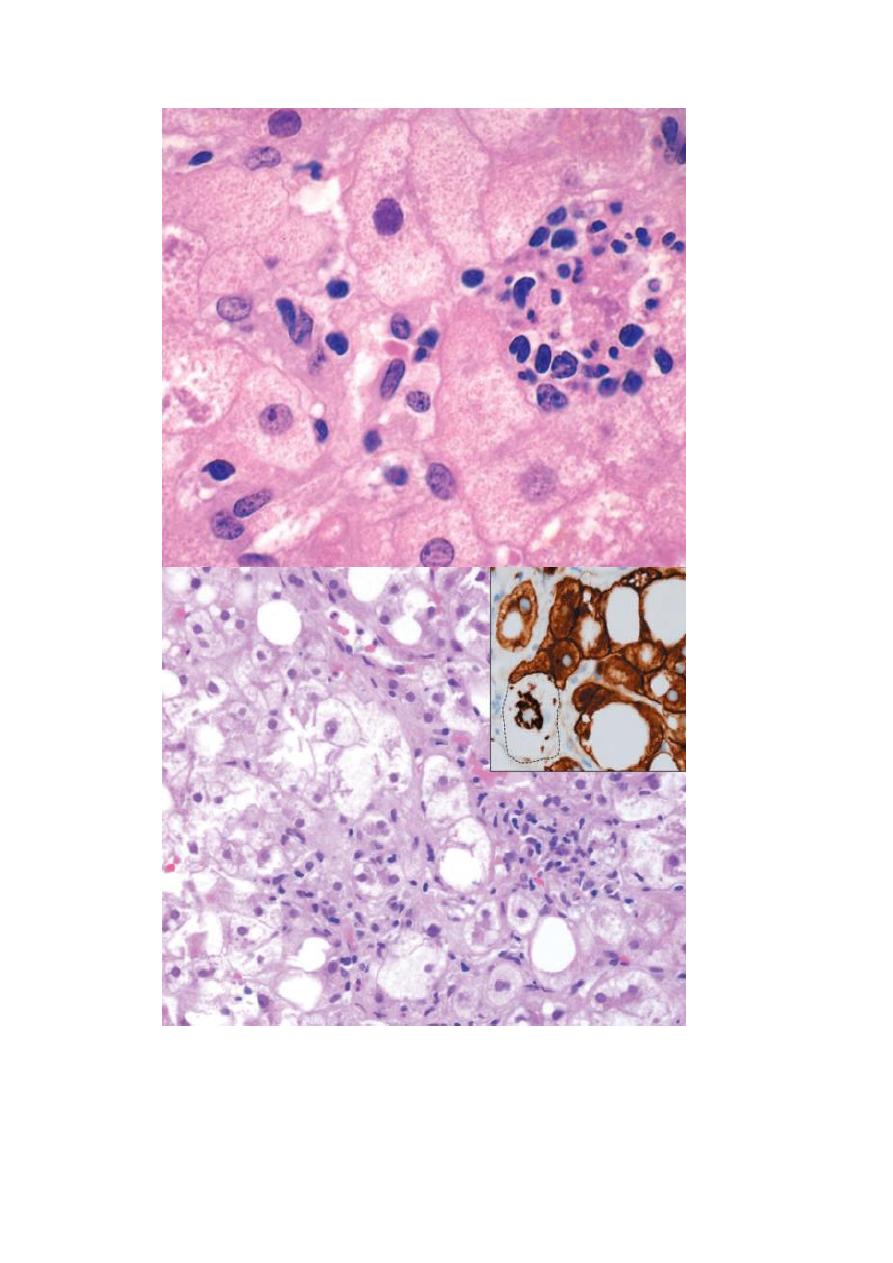
Fig. Hepatocyte injury in fatty liver disease associated with chronic alcohol use. (A) Clustered
inflammatory cells marking the site of a necrotic hepatocyte. A Mallory-Denk body is present in
another hepatocyte (arrow). (B) “Ballooned” hepatocytes (arrowheads) associated with clusters of
inflammatory cells. The inset stained for keratins 8 and 18 (brown) shows a ballooned cell (dotted
line) an immunoreactive Mallory-Denk body, leaving the cytoplasm “empty.”
7
3, Steatofibrosis. Fatty liver disease of all kinds has a distinctive pattern of scarring.
Like other changes, fibrosis appears first in the centrilobular region as central vein
sclerosis. fibrosis eventually link to portal tracts and then condense to create central
portal fibrous septa. As these become more prominent, the liver takes on a nodular,
cirrhotic appearance. Because in most cases the underlying cause persists, the
continual subdivision of established nodules by new, perisinusoidal scarring leads to
a classic micronodular cirrhosis . Early in the course, the liver is yellow-tan, fatty,
and enlarged, but with persistent damage over the course of years the liver is
transformed into a brown, shrunken, nonfatty organ composed of cirrhotic nodules
that are usually less than 0.3 cm in diameter—smaller than is typical for most chronic
viral
hepatitis.
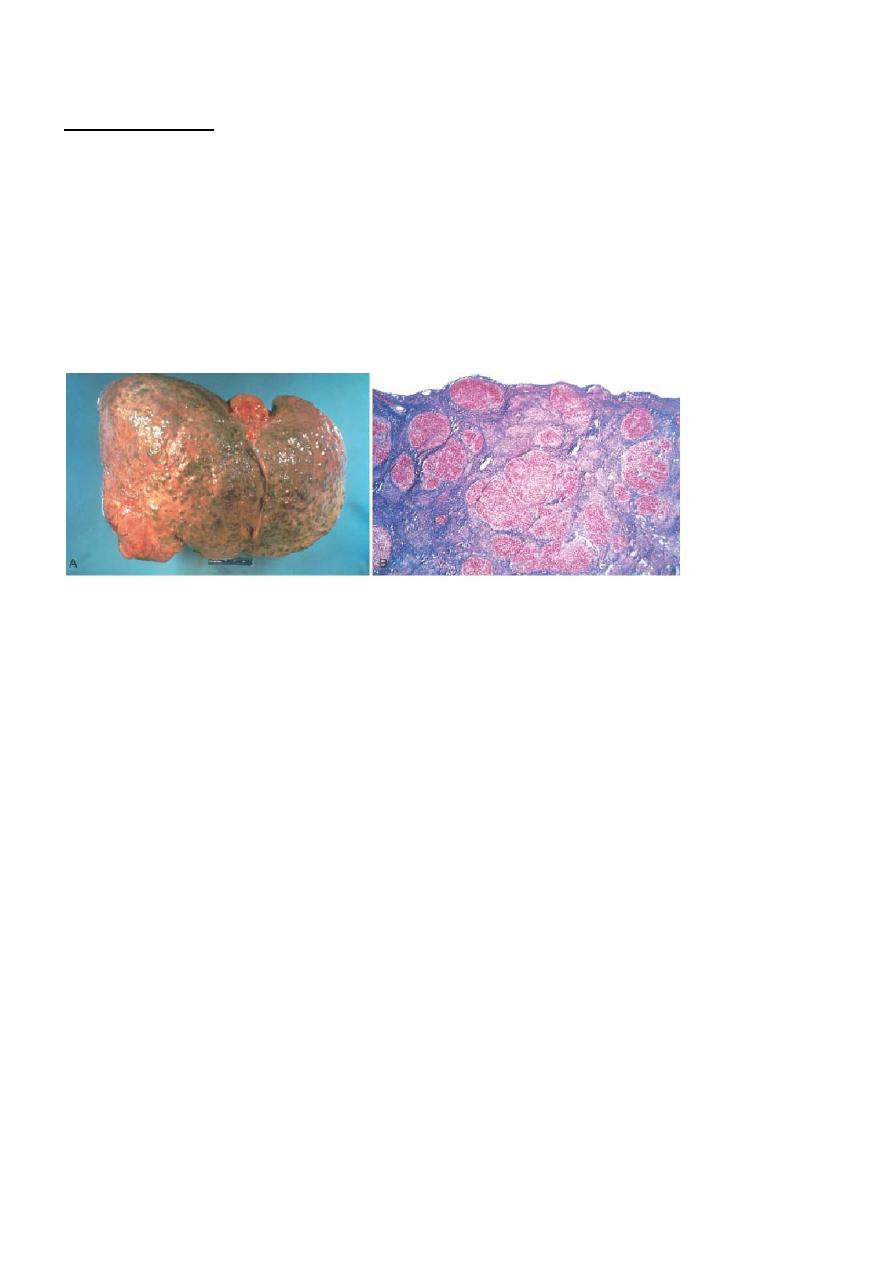
Alcoholic cirrhosis. A, The characteristic diffuse nodularity of the surface reflects the interplay between nodular
regeneration and scarring. The greenish tint of some nodules is due to bile stasis. A hepatocellular carcinoma is
present as a budding mass at the lower edge of the right lobe (lower left of figure). B, The microscopic view
shows nodules of varying sizes entrapped in blue-staining fibrous tissue. The liver capsule is at the top (Masson
trichrome).
Alcoholic Liver Disease
Between 90% and 100% of heavy drinkers develop fatty liver (i.e., hepatic steatosis),
and of those, 10% to 35% develop alcoholic hepatitis, whereas only 8% to 20% of
chronic alcoholics develop cirrhosis. Steatosis, alcoholic hepatitis, and fibrosis may
develop sequentially or independently, so they do not necessarily represent a
sequential continuum of changes. Hepatocellular carcinoma arises in 10% to 20% of
patients with alcoholic cirrhosis.
Pathogenesis of fatty liver:
Within hepatocyte, ethanol
1. increases fatty acids synthesis
2. decreases mitochondrial oxidation of fatty acids
3. increases the production of the triglycerides
4. impairs the release of lipoproteins.
The cause of alcoholic hepatitis is uncertain, but it may stem from one or
more of the toxic byproducts of ethanol and its metabolites.

8
Inhirited Metabolic Liver Diseases
1-
HEMOCHROMATOSIS
Hemochromatosis is characterized by the excessive accumulation of body
iron, most of which is deposited in parenchymal organs such as the liver
and pancreas. Iron can also accumulate in the heart, joints, or endocrine
organs. Hemochromatosis (also known as primary or hereditary
hemochromatosis) is a homozygous-recessive inherited disorder that is
caused by excessive iron absorption.
Accumulation of iron in tissues, which may occur as a consequence of
parenteral administration of iron, usually in the form of transfusions, or
other causes is variably known as secondary hemochromatosis.
the total body iron pool ranges from 2 to 6 gm in normal adults; about 0.5
gm is stored in the liver, 98% of which is in hepatocytes. In
hemochromatosis, total iron accumulation may exceed 50 gm, over one
third of which accumulates in the liver.
Pathogenesis.
Whatever the underlying cause, the onset of disease typically occurs after
20 gm of stored iron have accumulated. Excessive iron appears to be
directly toxic to host tissues. Mechanisms of liver injury include the
following:
• Lipid peroxidation via iron-catalyzed free radical reactions
• Stimulation of collagen formation by activation of hepatic stellate cells
• DNA damage by reactive oxygen species, leading to lethal cell injury or
predisposition to HCC
The deleterious effects of iron on cells that are not fatally injured are
reversible, and removal of excess iron with therapy promotes recovery of
tissue function.
The
morphologic
changes
in
severe
hemochromatosis
are
characterized
9
(1) tissue deposition of hemosiderin in the following organs (in decreasing
order of severity): liver, pancreas, myocardium, pituitary gland, adrenal
gland, thyroid and parathyroid glands, joints, and skin;
(2) cirrhosis; and (3) pancreatic fibrosis. In the liver, iron becomes evident
first as golden-yellow hemosiderin granules in the cytoplasm of periportal
hepatocytes, which can be histochemically stained with Prussian blue .
With increasing iron load, there is progressive deposition in the rest of the
lobule, the bile duct epithelium, and Kupffer cells. At this stage, the liver
typically is slightly enlarged and chocolate brown. Fibrous septa develop
slowly, linking portal tracts to each other and leading ultimately to
cirrhosis in an intensely pigmented (very dark brown to black) liver.
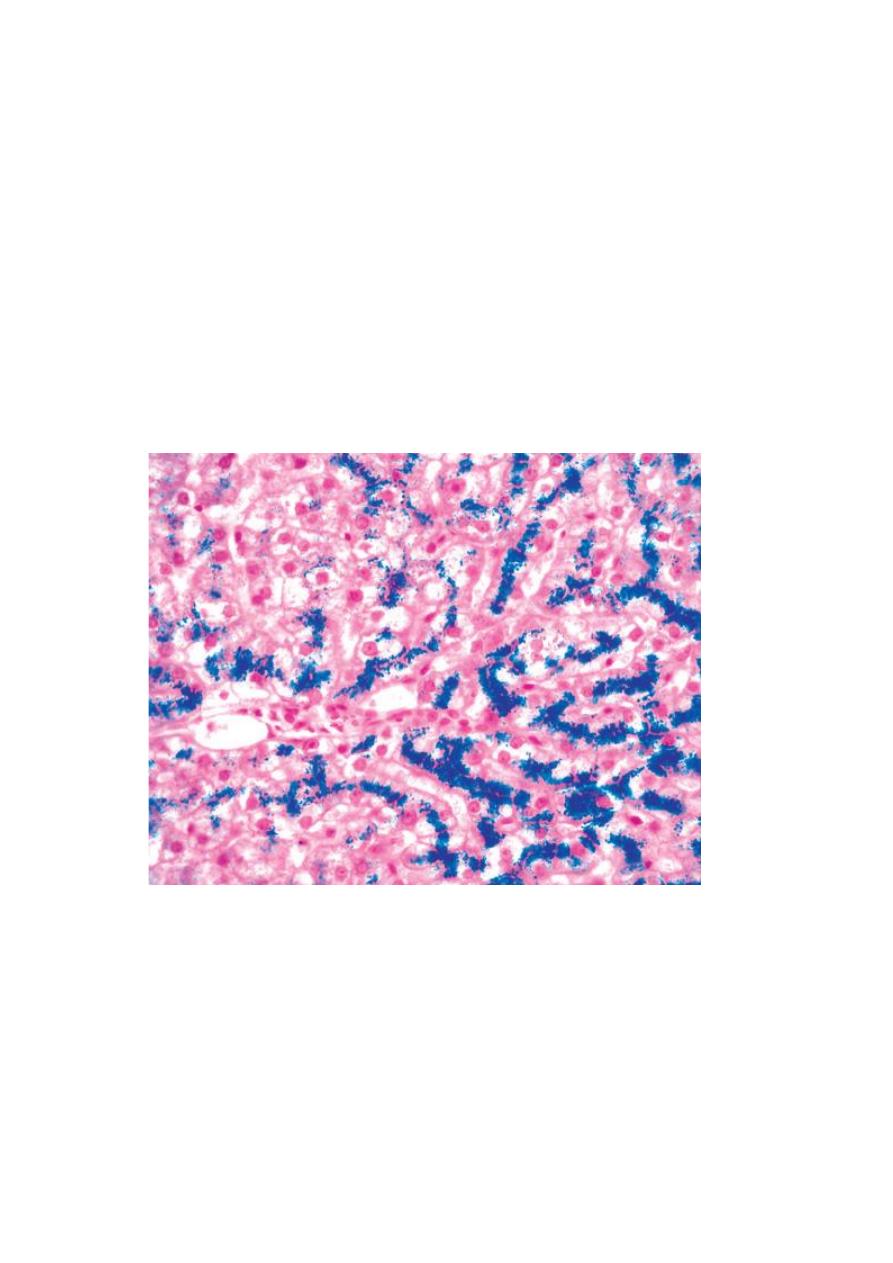
Fig. Hereditary hemochromatosis. In this Prussian blue–stained section, hepatocellular iron appears blue.
The parenchymal architecture is normal at this stage of disease, even with such abundant iron.
WILSON DISEASE
Wilson disease is an autosomal recessive disorder caused by mutation of the ATP7B
gene (located on chromosome 13, encodes a transmembrane copper-transporting
ATPase, expressed on the hepatocyte canalicular membrane.), resulting in impaired
copper excretion into bile and a failure to incorporate copper into ceruloplasmin. This
disorder is marked by the accumulation of toxic levels of copper in many tissues and

10
organs, principally the liver, brain, and eye. Normally, 40% to 60% of ingested
copper is absorbed in the duodenum and proximal small intestine, and is transported
to the portal circulation complexed with albumin and histidine. Free copper
dissociates and is taken up by hepatocytes. Copper is incorporated into enzymes and
also binds to a α2-globulin (apoceruloplasmin) to form ceruloplasmin, which is
secreted into the blood. Excess copper is transported into the bile. Ceruloplasmin
accounts for 90% to 95% of plasma copper. Circulating ceruloplasmin is eventually
degraded within lysosomes, after which the released copper is excreted into bile. This
degradation/excretion pathway is the primary route for copper elimination.
Deficiency in the ATP7B protein causes a decrease in copper transport into bile,
impairs its incorporation into ceruloplasmin, and inhibits ceruloplasmin secretion into
the blood. These changes cause copper accumulation in the liver and a decrease in
circulating ceruloplasmin. The copper causes toxic liver injury.
Morphology. The liver often bears the brunt of injury, but the disease may also
present as a neurologic disorder. The hepatic changes are variable, ranging from
relatively minor to massive damage. Fatty change (steatosis) may be mild to
moderate. An acute hepatitis can show features mimicking acute viral hepatitis,
except possibly for the accompanying fatty change. The chronic hepatitis of Wilson
disease exhibits moderate to severe inflammation and hepatocyte necrosis, with the
particular features of macrovesicular steatosis and Mallory bodies. With progression
of chronic hepatitis, cirrhosis will develop.
In the brain, toxic injury primarily affects the basal ganglia, particularly the putamen,
which shows atrophy and even cavitation. Nearly all patients with neurologic
involvement develop eye lesions called Kayser-Fleischer rings, green to brown
deposits of copper in Desçemet's membrane in the limbus of the cornea.