
Platelet Disorders
TUCOM
Dep. of Medicine
5
th
year
Dr. Hasan I. Sultan
8-1-2019

Platelet disorders
Learning objectives:
1. Review the function of platelets
2. Enumerate the causes, investigations and management
of thrombocytopenia
3. Explain the following conditions: idiopathic
thrombocytopenic purpura, thrombotic
thrombocytopenic purpura, heparin-induced
thrombocytopenia and disseminated intravascular
coagulation
4. Clarify the platelet function disorders
5. Enumerate the causes of thrombocytosis
6. Show the causes of non-thrombocytopenic purpura

Platelets
Platelets are formed in the bone marrow from
megakaryocytes. The formation and maturation of
megakaryocytes are stimulated by thrombopoietin,
which produced in the liver. When platelets are released
into the circulation, they survive between 7 to 10 days.
Some 30% of peripheral platelets are normally pooled in
the spleen and do not circulate.
Platelets are anucleate cells, discoid in shape, with a
diameter of 2- 4 μm. The surface membrane invaginates
to form a tubular network, the canalicular system, which
provides a conduit for the discharge of the granule
content following platelet activation.
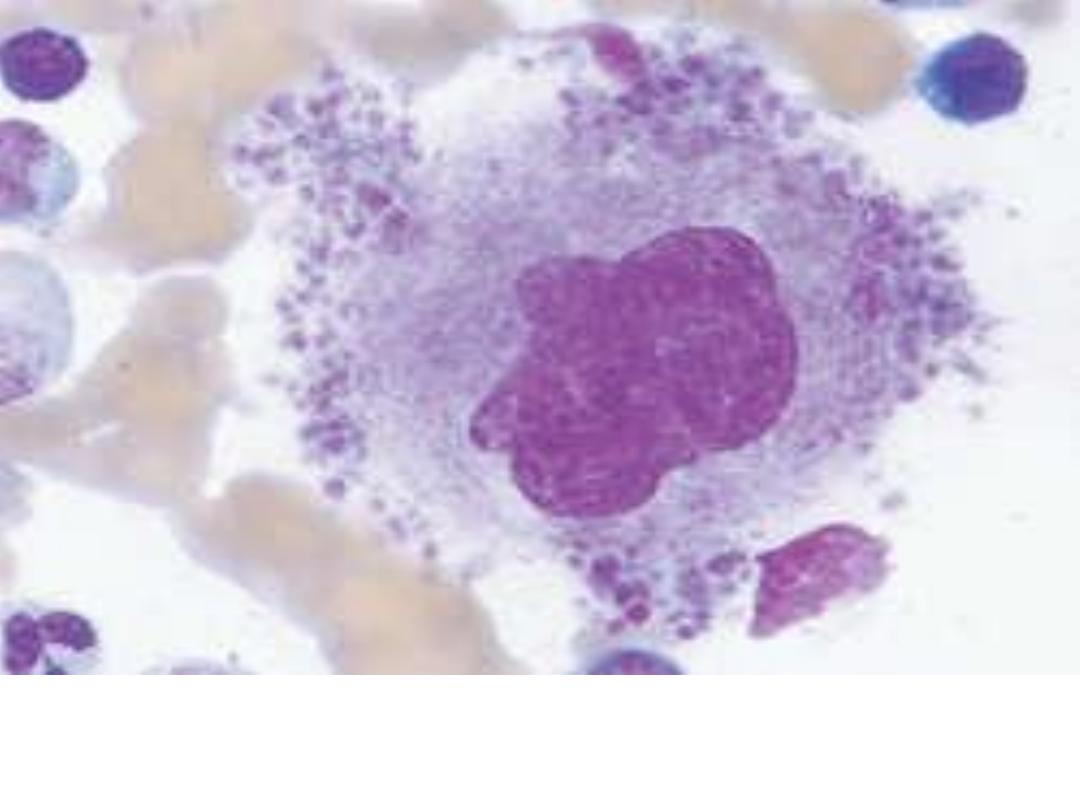
Megakaryocyte: Large bone marrow cell with large or
multiple nuclei from which platelets are derived.
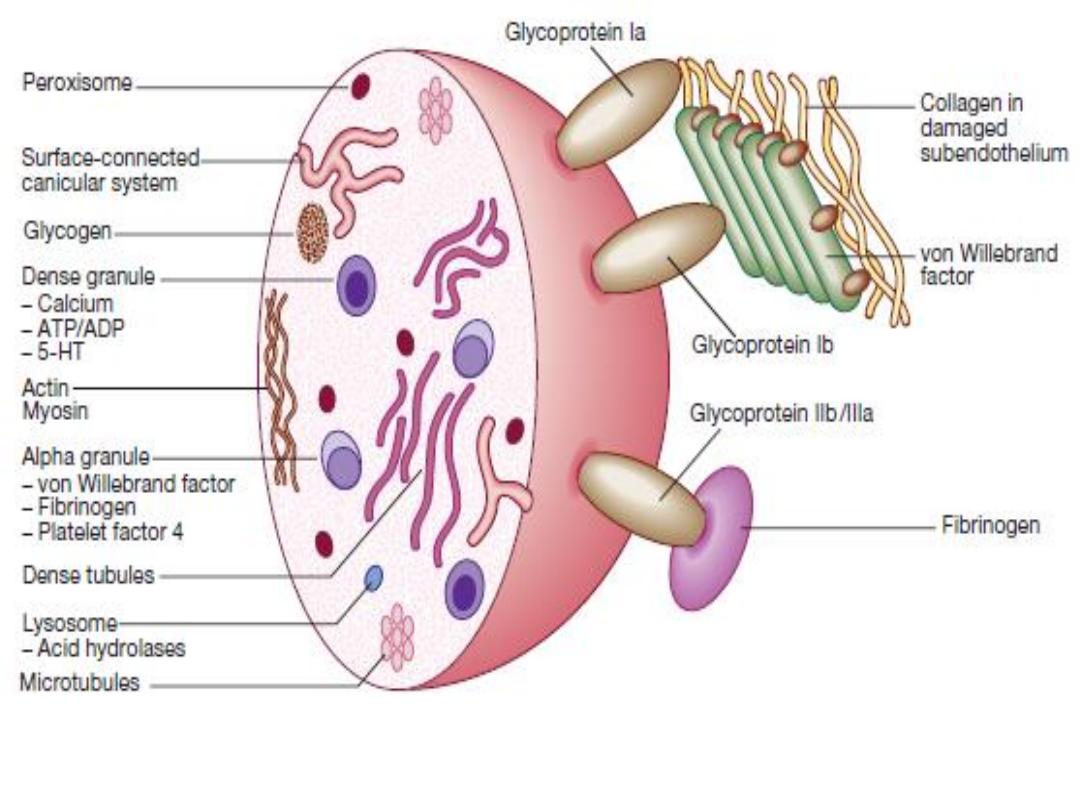
Normal platelet structure. (5 HT = 5-hydroxytryptamine, serotonin;
ADP = adenosine diphosphate; ATP = adenosine triphosphate
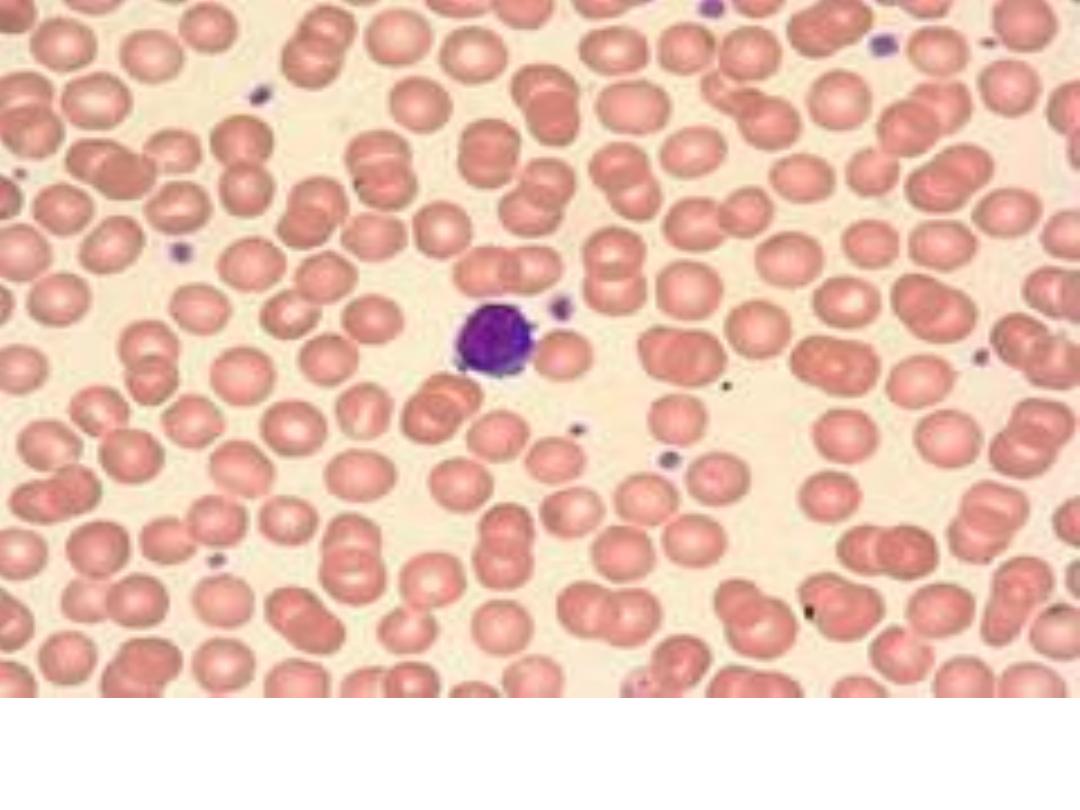
Normal Peripheral Smear: normal red cells, a lymphocyte
and platelets

The platelet functions as the cellular-based platform
for hemostasis. Platelets mediate primary
hemostasis through:
1- Receptors adhesion activation: by binding to
endothelium and subendothelium at sites of damage
• GPIb/IX/V-vWF
• GPIIb/IIIa-fibrinogen and GPIIb/IIIa-vWF
• GPIa/IIa-collagen
• GPV-thrombin
2- Secreted α-Granule Proteins: fibrinogen, vWF, α2-
antiplasmin, factors V, VIII, and XI, Antiheparin
(platelet factor 4)
3- Secreted Dense-Granule Agonists: ADP, serotonin
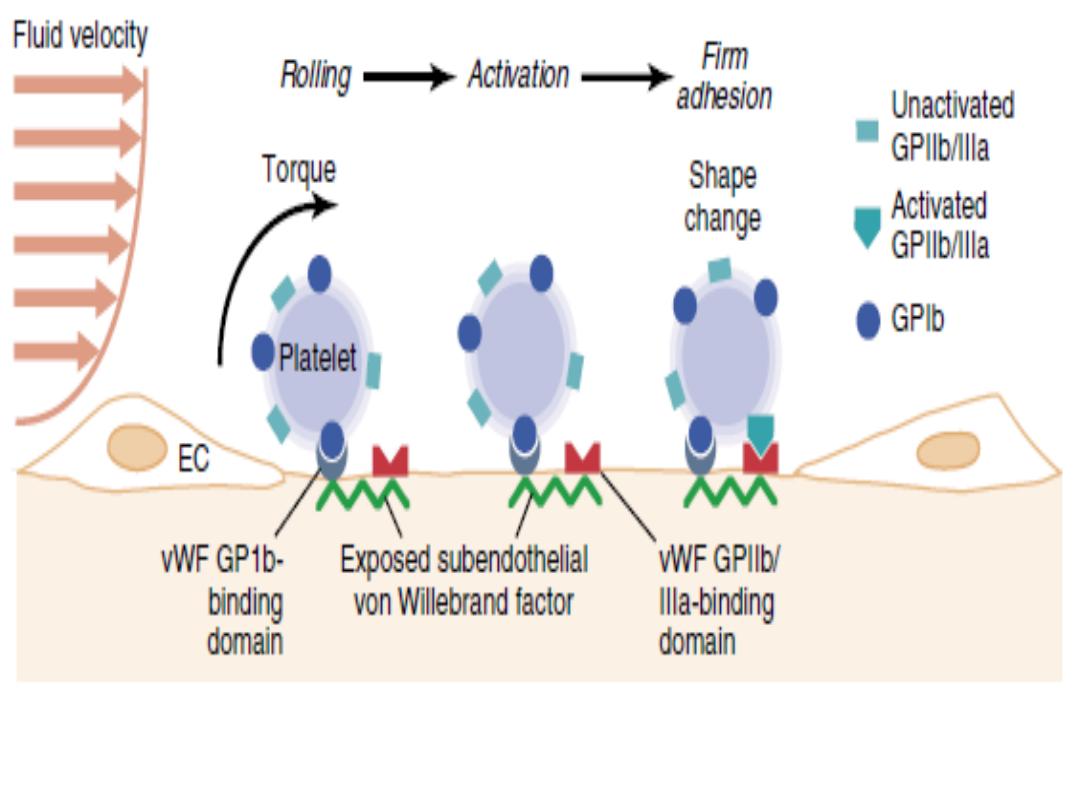
The adhesive interactions producing stable platelet
attachment to subendothelial von Willebrand factor (vWF).

The normal platelet count range is between 150,000-
450,000 per mm³ (or microlitre) or 150 - 450 x 10
9
/L.
With platelet counts in the normal range and normal
platelet function, the bleeding time, is generally less
than 8 minutes.

Thrombocytopenia (low platelet count)
Thrombocytopenia (platelet count <150 x 10
9
/L) is one of
the most common problems in hospitalized patients.
Spontaneous bleeding does not usually occur until the
platelet count falls below 20 × 10
9
/L.
Patterns of spontaneous bleeding due to thrombocytopenia:
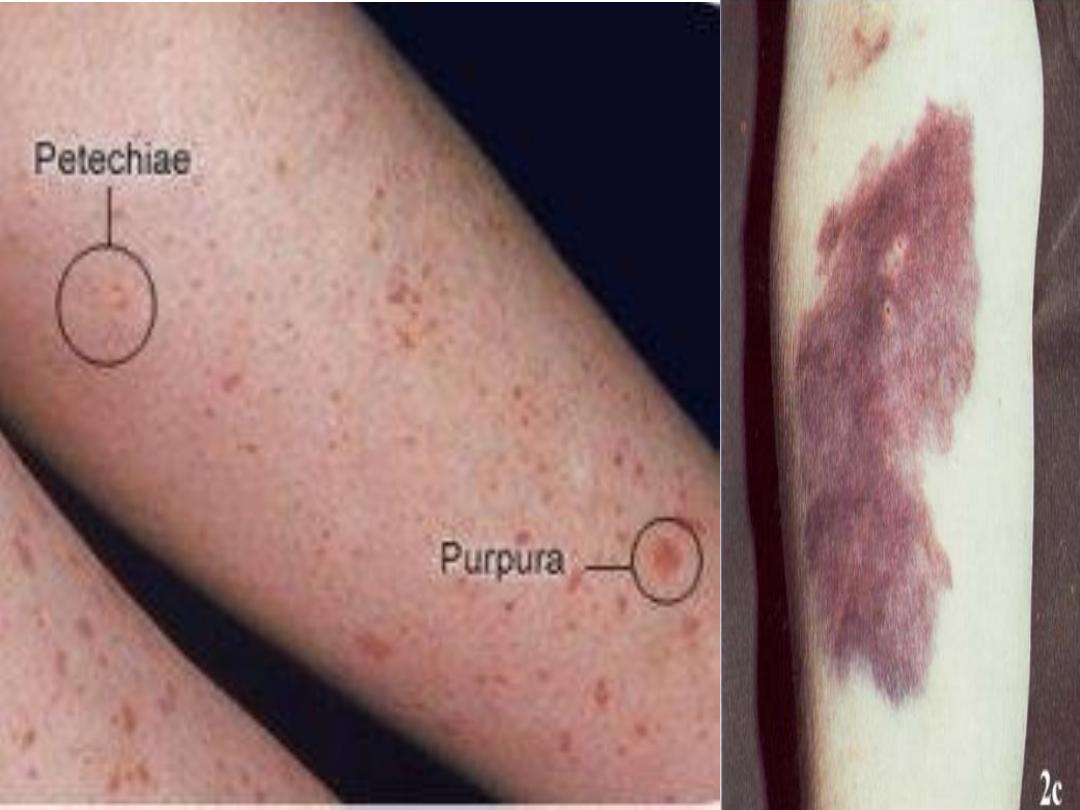
Petechiae: Pinpoint or 1 mm hemorrhages. Purpura: larger
(> 3 mm) hemorrhages. Ecchymoses: larger (> 2 cm)
hemorrhages. Petechiae, purpura and ecchymoses are not
palpable and not blanch when pressed are characteristic,
but there may also be oral, nasal, gastrointestinal or
genitourinary bleeding.
Severe thrombocytopenia (< 10 × 10
9
/L) may result in retinal
haemorrhage and potentially fatal intracranial bleeding, but
this is rare.

Causes of thrombocytopenia
A- Decreased production
1- Marrow hypoplasia:
Fanconi’s anaemia. Idiopathic
aplastic anaemia. Drug-induced: cytotoxics, antimetabolites.
Transfusion-associated graft-versus-host disease.
2- Marrow infiltration:
Leukaemia. Myeloma. Carcinoma
(rare). Myelofibrosis. Osteopetrosis. Lysosomal storage
disorders, e.g. Gaucher’s disease.
3- Haematinic deficiency:
Vitamin B12 and/or folate
deficiency
4- Familial (macro-) thrombocytopathies:
Alport’s syndrome.
Bernard Soulier disease. Wiskott–Aldrich syndrome.

B- Increased consumption
1- Immune mechanisms
: Idiopathic thrombocytopenic
purpura. Post-transfusion purpura. Drug-associated,
especially quinine and vancomycin. Heparin-induced
thrombocytopenia.
2- Coagulation activation:
Disseminated intravascular
coagulation.
3- Mechanical pooling:
Hypersplenism
4- Thrombotic microangiopathies:
Haemolytic uraemic
Syndrome. Liver disease. Thrombotic thrombocytopenic
purpura. Pre-eclampsia

Investigations:
• Are directed at the possible causes.
• A blood film is the single most useful initial
investigation.
• Examination of the bone marrow may reveal
increased megakaryocytes in consumptive causes of
thrombocytopenia, or the underlying cause of bone
marrow failure in leukaemia, hypoplastic anaemia or
myelodysplasia.
Treatment:
(if required) depends on the underlying
cause.
Platelet transfusion is rarely required and is usually
confined to patients with bone marrow failure and
platelet counts below 10 × 10
9
/L, or to clinical situations
with actual or predicted serious haemorrhage.

Idiopathic thrombocytopenic purpura (ITP)
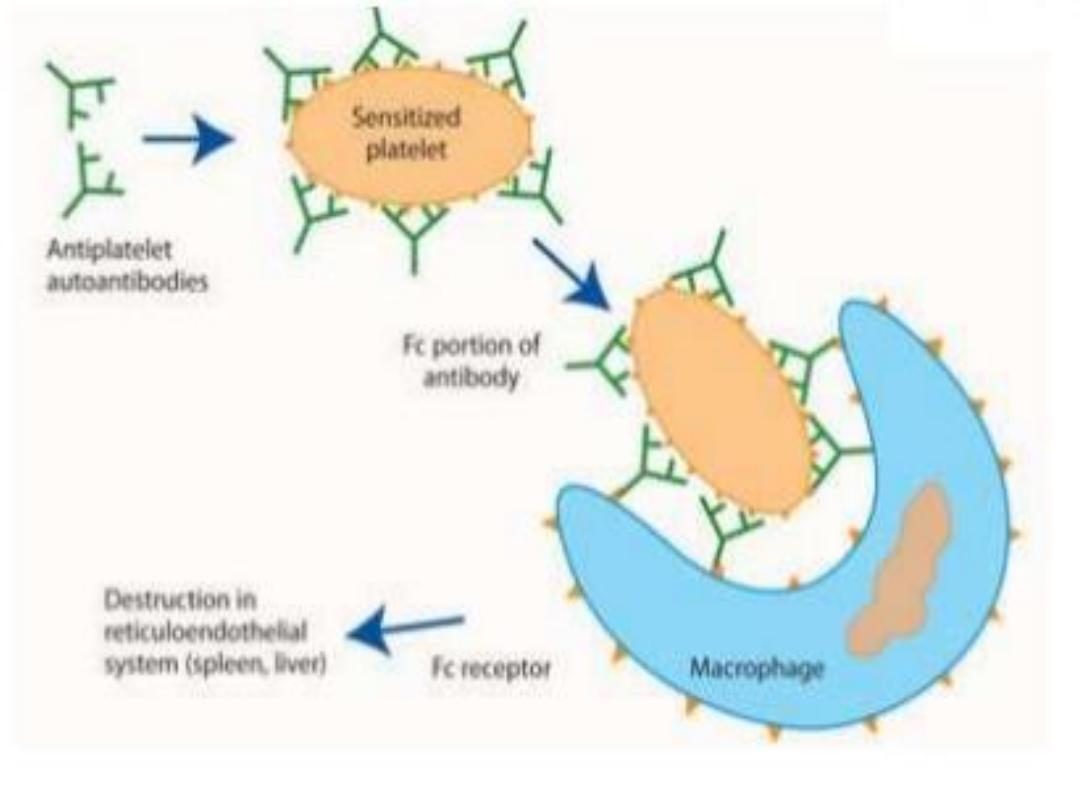
ITP: Is an immune mediated by autoantibodies, most often
directed against the platelet membrane glycoprotein IIb/IIIa,
which sensitise the platelet, resulting in premature removal
from the circulation by cells of the reticulo-endothelial
system.
It is either occur in isolation or associated with underlying
immune dysregulation such as connective tissue diseases,
HIV infection, B cell malignancies and pregnancy.
Clinical features:
In children, ITP is usually acute often preceded by a viral
infection, such as varicella. In adults, ITP more commonly
affects females, an insidious onset and it is unusual for
preceding viral infection.

The presentation depends on the degree of
thrombocytopenia.
Spontaneous bleeding typically occurs only when the
platelet count is below 20 × 10
9
/L.
At higher counts, the patient may complain of easy bruising
or sometimes epistaxis or menorrhagia.
Many cases with counts of more than 50 × 10
9
/L are
discovered by chance.
Investigations:
The diagnosis of ITP is partly made by exclusion. Fever,
organomegaly, pancytopenia, lymphadenopathy, or
abnormal peripheral blood cells should prompt an
evaluation for malignant disease, such as leukemia, other
bone marrow disorders.

• The blood smear is normal, apart from a greatly
reduced platelet number, but no other abnormal
cells such as blasts, which would accompany
leukemia.
• The bone marrow demonstrates an obvious
increase in megakaryocytes.
• Autoantibody testing performed if a diagnosis of
connective tissue disease is likely.
• HIV testing should be considered.
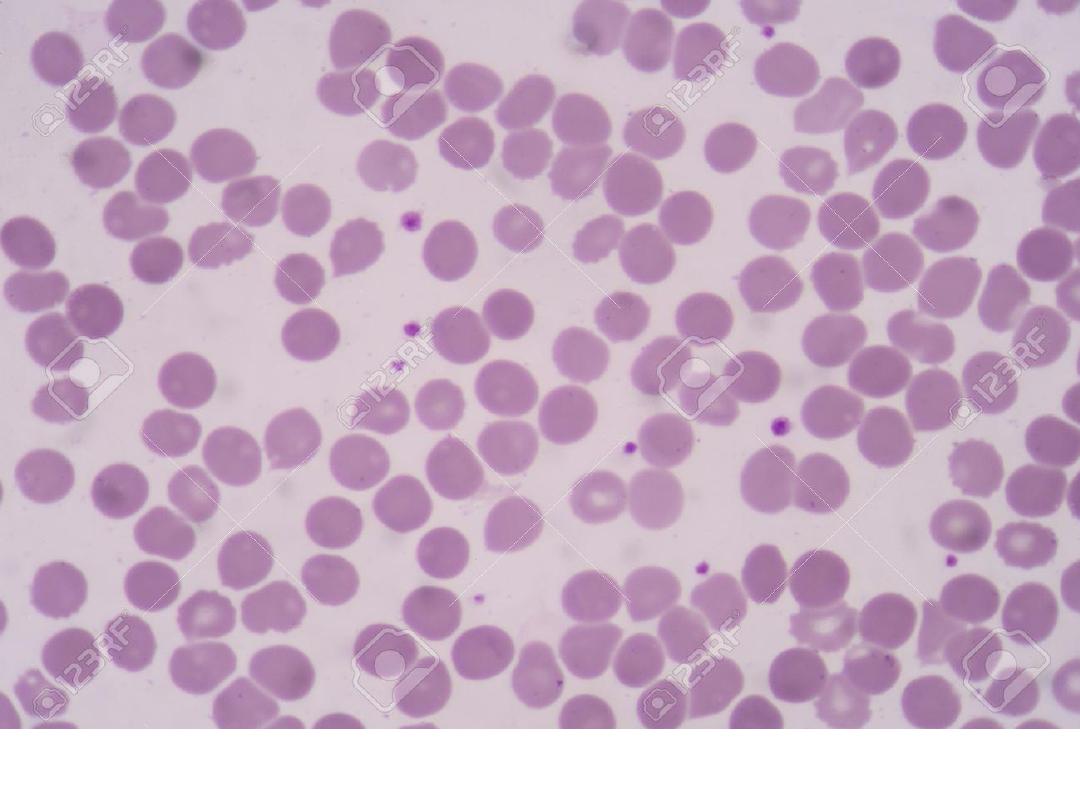
Adequate platelet count in blood smear
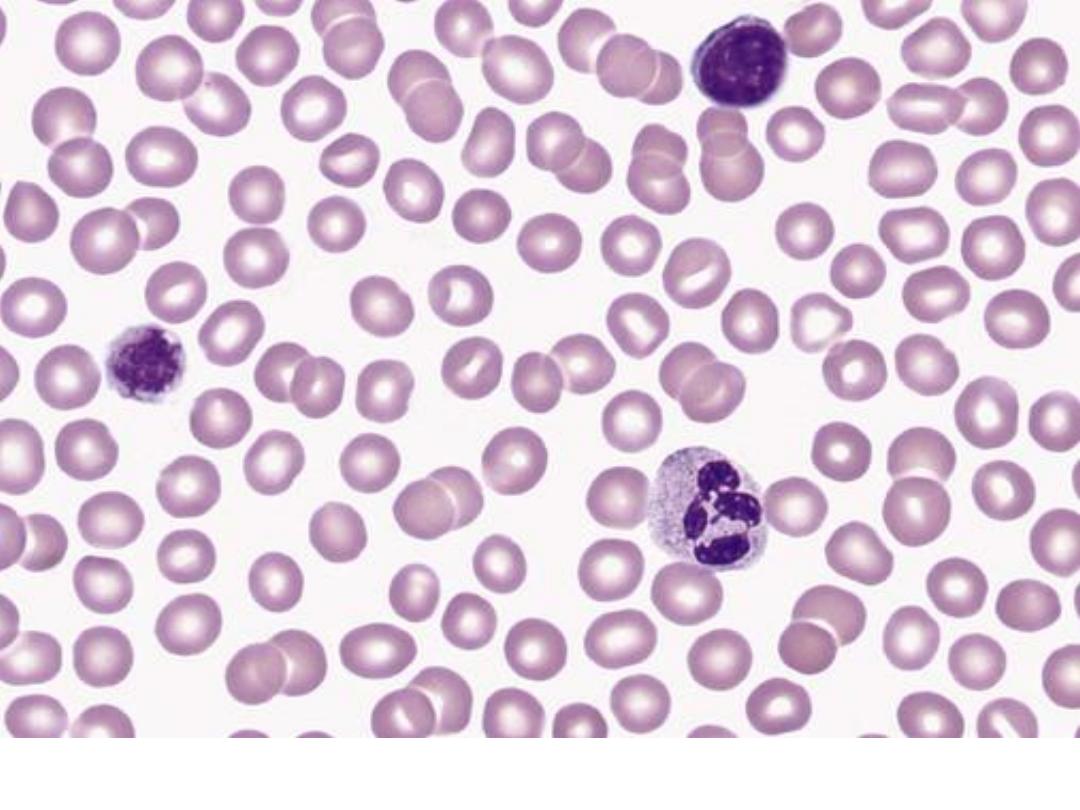
ITP: Review of the peripheral smear reveals a paucity of platelets

Management
• Stable compensated ITP and a platelet count of more than
30 ×
10
9
/L do not require treatment to raise the platelet
count, except at times of increased bleeding risk, such as
surgery and biopsy.
• First-line therapy for patients with spontaneous bleeding is
with prednisolone 1 mg/kg daily to suppress antibody
production and inhibit phagocytosis of sensitised platelets
by reticuloendothelial cells.
• Administration of intravenous immunoglobulin (IVIg) can
raise the platelet count by blocking antibody receptors on
reticuloendothelial cells, and is combined with
corticosteroid therapy if there is severe haemostatic failure
or a slow response to steroids alone.
• Life threatening bleeding should be treated with platelet
transfusion in addition to the other therapies

• Relapses should be treated by re-introducing
glucocorticoids.
• If a patient has two relapses or primary refractory
disease, second-line therapies are considered,
which include the thrombopoietin receptor
agonists eltrombopag and romiplostim,
splenectomy and immunosuppression.
• Low-dose corticosteroid therapy,
immunosuppressants such as rituximab,
ciclosporin and tacrolimus should be considered in
cases where the approaches above are ineffective.

Thrombotic thrombocytopenic purpura (TTP)
TTP is a disorder in which thrombosis is accompanied by
paradoxical thrombocytopenia. TTP is characterized by a
pentad of findings, although few patients have all five
components:
1. Thrombocytopenia
2. Microangiopathic haemolytic anaemia
3. Neurological sequelae
4. Fever
5. Renal impairment
It is an acute autoimmune disorder mediated by antibodies
against ADAMTS-13 (a disintegrin and metalloproteinase
with a thrombospondin type-1 motif).
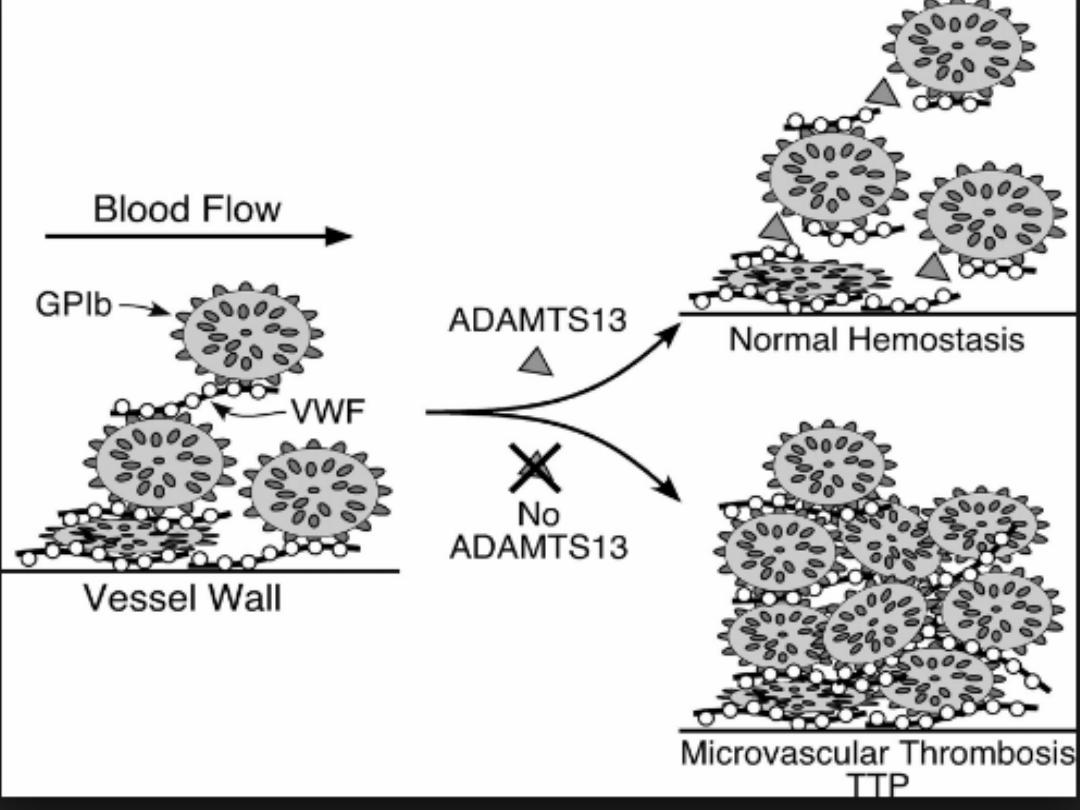

This enzyme normally cleaves vWF multimers to
produce normal functional units, and its deficiency
results in large vWF multimers which cross-link
platelets.
The features are of microvascular occlusion by platelet
thrombi affecting key organs, principally brain and
kidneys.
It is a rare disorder (1 in 750 000 per annum), which may
occur alone or in association with drugs (ticlopidine,
ciclosporin), HIV, shiga toxins and malignancy.
It should be treated by emergency plasma exchange.
Corticosteroids, aspirin and rituximab also have a role in
management. Untreated mortality rates are 90% in the
first 10 days, and even with appropriate therapy, the
mortality rate is 20–30% at 6 months.

Heparin-induced thrombocytopenia (HIT)
HIT is a rare complication of heparin therapy, caused by
induction of anti-heparin/PF4 antibodies which bind to and
activate platelets via an Fc receptor.
This results in platelet activation and a prothrombotic state,
with a paradoxical thrombocytopenia. HIT is more common
with use of UFH rather than LMWH, and with higher doses
of heparin.
Clinical features
Patients present, typically 5–14 days after starting heparin
treatment. They may be asymptomatic, or develop
venous or arterial thrombosis and skin lesions, including
overt skin necrosis.
Diagnosis
is by using the 4Ts scoring system:
1. The thrombocytopenia
2. The timing of the fall in platelet count
3. The presence of new thrombosis
4. The likelihood of another cause for the
thrombocytopenia
.
Management
Heparin should be discontinued as soon as HIT is diagnosed
and an alternative anticoagulant which does not cross-react
with the antibody substituted. Argatroban (a direct
thrombin inhibitor) and danaparoid (a heparin analogue)

Disseminated intravascular coagulation (DIC)
DIC is a life-threatening nonimmune platelet destruction,
characterised by systemic activation of the pathways
involved in coagulation and its regulation. This may result in
the generation of intravascular fibrin clots causing
multiorgan failure, with simultaneous coagulation factor and
platelet consumption causing bleeding. This triggered by
endotoxin release or by severe tissue damage.
There is consumption of platelets, coagulation factors
(notably factors V and VIII) and fibrinogen. The lysis of fibrin
clot results in production of fibrin degradation products
(FDPs), including D-dimers.


Management of DIC:
• Treat the underlying cause.
• Admission to intensive care to deal with
concomitant issues, such as acidosis, dehydration,
renal failure and hypoxia.
• Blood component therapy, such as fresh frozen
plasma, cryoprecipitate and platelets, should be
given if the patient is bleeding
• Established thrombosis should be treated
cautiously with therapeutic doses of
unfractionated heparin, unless clearly
contraindicated.
Platelet function disorders
Causes
A- Acquired platelet function abnormalities
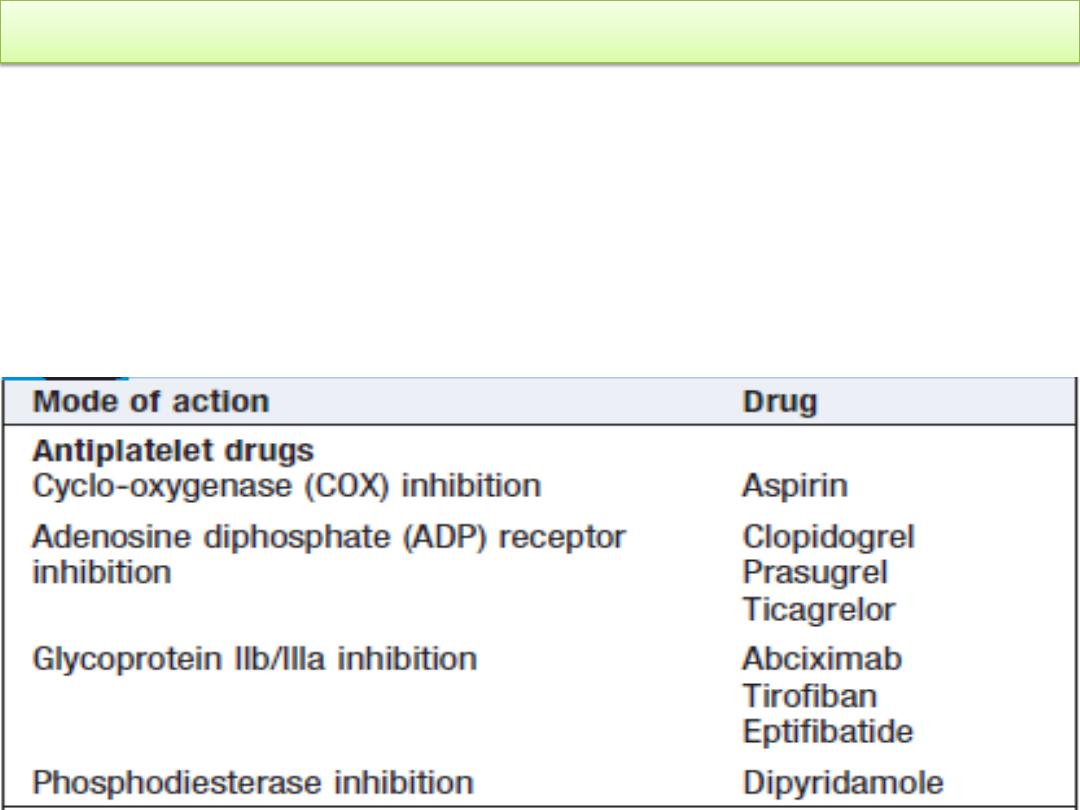
1- Drugs: The most common acquired disorders are iatrogenic,
resulting from the use of aspirin, clopidogrel, dipyridamole and
the GP IIb/IIIa inhibitors to prevent arterial thrombosis.
Modes of action of antiplatelet drugs:

2- Uremic platelet dysfunction is caused by toxic proteins
that accumulate in renal failure that inhibits platelet
function. Control of uremic bleeding by:
1. Dialysis
2. Maintenance of the hematocrit
3. DDAVP and cryoprecipitate, which has been shown to
shorten the bleeding time significantly.
4. Conjugated estrogens are of some benefit for long-term
treatment.
5. Platelet transfusions may be useful in patients with life
threatening bleeding.

B- Inherited platelet function abnormalities are relatively
rare
1- Deficiency of the membrane glycoproteins, e.g. Bernard-
Soulier syndrome and Glanzmann thrombasthenia.
Bernard-Soulier syndrome is caused by decreased surface
expression of platelet GPIb (the primary von Willebrand
factor) characterized by mild thrombocytopenia, increased
bleeding time, and mild-to moderate bleeding symptoms.
Glanzmann thrombasthenia is characterized by an increased
bleeding time and abnormally low levels of expression of
platelet GPIIb/IIIa (the receptor for both vWF and
fibrinogen) is an autosomal recessive condition associated
with a variable but often severe bleeding disorder.

2- Defective plateletn granules, e.g. a deficiency of dense
(delta) granules, giving rise to storage pool disorders
3- Congenital macrothrombocytopathies that are due to
mutations in the myosin heavy chain gene MYH-9 are
characterised by large platelets, inclusion bodies in the
neutrophils (Döhle bodies) and a variety of other features,
including sensorineural deafness and renal abnormalities.
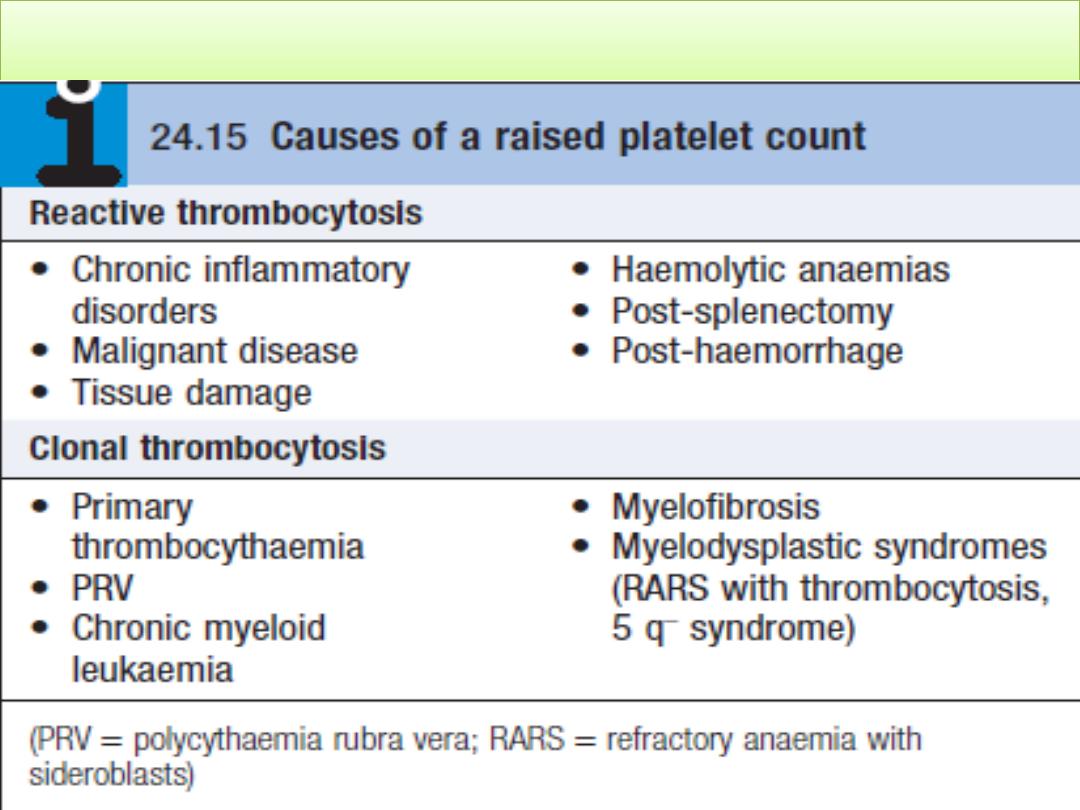
Thrombocytosis (high platelet count)

• Is the presence of high platelet counts in the blood , it mean
more than 450,000 per mm³ (or microlitre) (or 450 x 10
9
/L)
• The most common reason for a raised platelet count is that it
is reactive to another process such as infection, connective
tissue disease, malignancy, iron deficiency, acute haemolysis
or gastrointestinal bleeding
• Patients with PRV, essential thrombocythaemia and
occasionally myelofibrosis may present with thrombosis.
Stroke and transient ischaemic attacks, amaurosis fugax, and
digital ischaemia or gangrene are also features. In addition, to
itching after exposure to water (aquagenic pruritus),
splenomegaly and systemic upset. Platelet counts greater than
1,000,000/mcL are thought to increase the risk for thrombosis.
Aspirin is indicated only in patients with symptomatic
thrombosis. Concomitant therapy to prevent thrombotic
complications of thrombocytosis includes lowering the
platelet count with hydroxyurea.
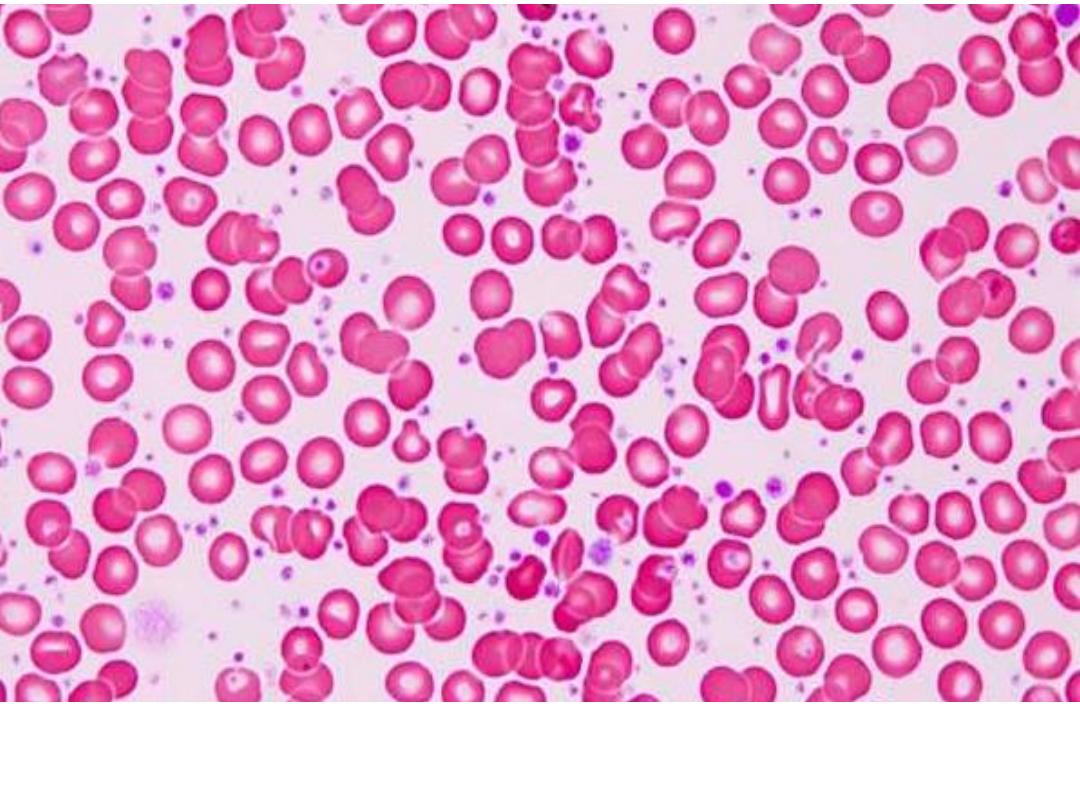
Peripheral blood smear in essential thrombocytosis showing increased
platelet numbers.

Causes of non-thrombocytopenic purpura
1. Senile purpura
2. Factitious purpura
3. Henoch- Schönlein purpura
4. Vasculitis
5. Paraproteinaemias
6. Purpura fulminans, e.g. in DIC secondary to sepsis
Petechiae / purpura is minor bleeding into the dermis that is
flat and non-blanching. Petechiae are typically found in
patients with thrombocytopenia or platelet dysfunction.
Palpable purpura occurs in vasculitis.
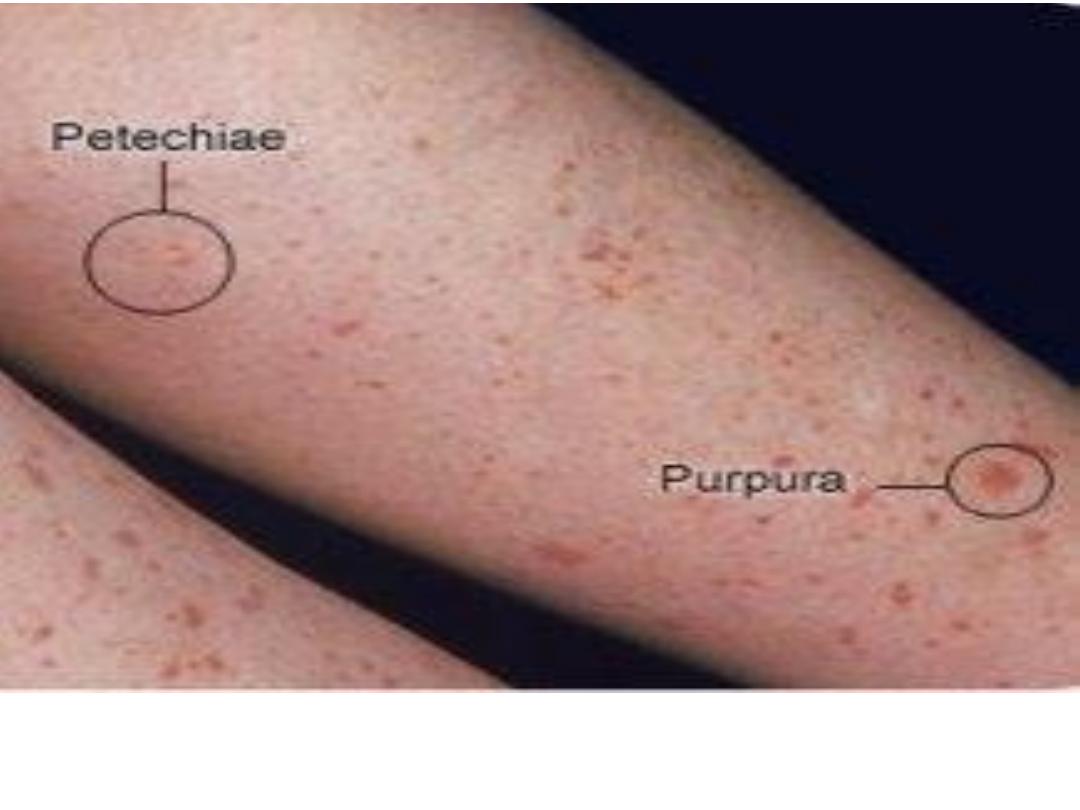
Pinpoint red spots on the skin, called petechiae with larger purplish
areas (purpura) caused by bleeding under the skin in patient with ITP
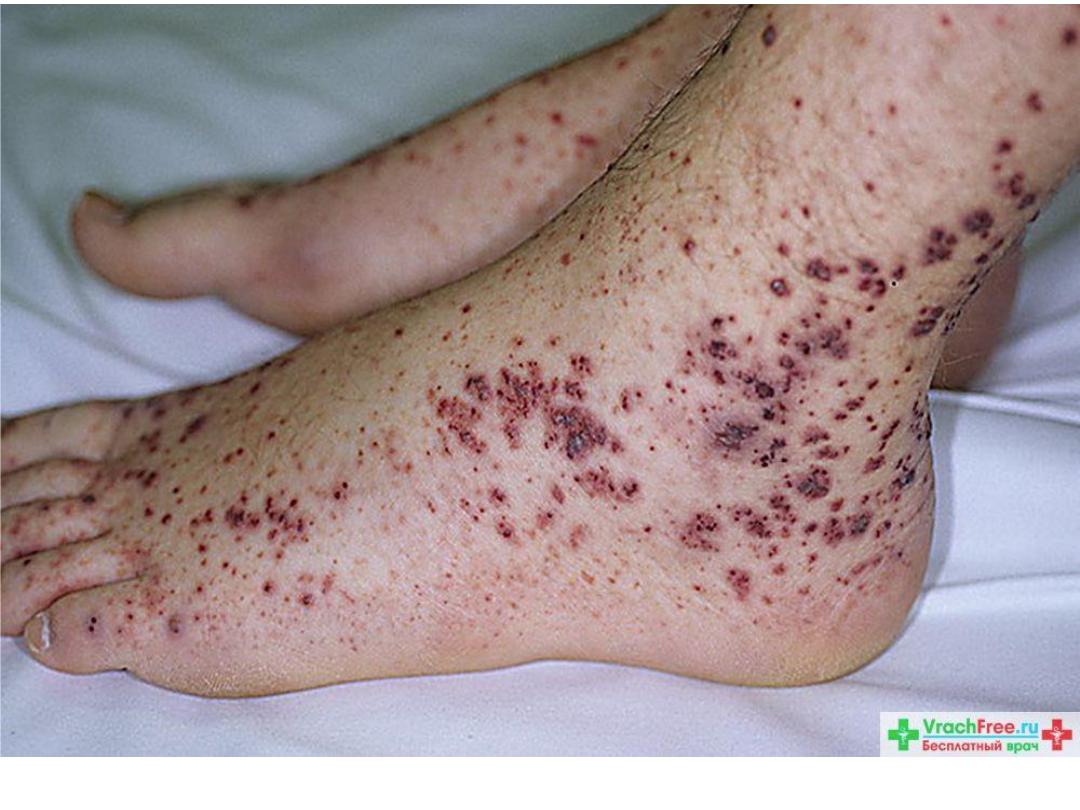
Vasculitis , Henoch - Schonlein purpura

Thanks