Dr. Mohamed Ghalib
Internal Medicine
TUCOM
5
th
year
Aplastic Anemia

Aplastic anemia (AA) is a rare disorder characterized by
pancytopenia with a markedly hypocellular bone marrow. This
disease was first described in 1888 by Paul Ehrlich, who
observed that autopsy bone marrow specimens from a young
woman who died of severe anemia and neutropenia were
extremely hypoplastic.
Later studies demonstrated that patients with severe AA
possessed only a fraction of normal pluripotent stem cell
numbers despite normal functional marrow stromal cells and
normal or even elevated levels of stimulatory cytokines.

The incidence of AA ranges from 1 to 5 cases per million people in the
general population.
It occurs predominantly in young adults (20 to 25 years old) and older
adults (60 to 65 years old).
The incidence is threefold higher in developing countries (e.g., Thailand
and China) compared with industrialized Western nations (e.g., Europe
and USA), a fact that is not explained by differences in drug or radiation
exposure.
A few AA cases occur in the context of a congenital bone marrow failure
disorder, such as Fanconi’s anemia, Shwachman-Diamond syndrome, and
dyskeratosis congenita.
The most common congenital AA, Fanconi’s anemia, is an autosomal
recessive disorder arising from mutations in genes encoding DNA repair
proteins.

Primary idiopathic acquired aplastic
anaemia
This is a rare disorder in Europe and North America, with 2–4 new cases
per million population per annum. The disease is much more common in
certain other parts of the world, e.g. east Asia. The basic problem is
failure of the pluripotent stem cells because of an autoimmune attack,
producing hypoplasia of the bone marrow with a pancytopenia in the
blood. The diagnosis rests on exclusion of other causes of secondary
aplastic anaemia and rare congenital causes, such as Fanconi’s anaemia.

Secondary aplastic anaemia
That is occur secondary to ecposure to an offending drug or
chemical.
It is important to check the reported side-effects of all drugs taken
over the preceding months.
In some instances, the cytopenia is more selective and affects only
one cell line, most often the neutrophils. Frequently, this is an
incidental finding, with no ill health. It probably has an immune
basis but this is difficult to prove.


Pathology
The known causes of acquired AA are numerous and range from
myeloablative radiation exposure to common viruses and
medications. Prior bone marrow toxicity from drugs, chemicals
(e.g., benzene, cyclic hydrocarbons found in petroleum products,
rubber glue, insecticides, chemical dyes), or radiation
predisposes to AA because these agents directly injure
proliferating and differentiating HSCs by inducing DNA damage.
In contrast, cytotoxic chemotherapy (especially with alkylating
agents) and radiation therapy target all rapidly cycling cells and
often induce reversible bone marrow aplasia.
Despite the many causes of acquired AA, most cases are
idiopathic.

Acquired and congenital AAs appear to be etiologically linked through
abnormal telomere maintenance. Telomeres are repeated nucleotide
sequences that cap and protect chromosome ends from degradation. Cell
division leads to normal telomere erosion;
when telomeres reach a critically short length, cells cease to proliferate,
senesce, and undergo apoptosis, often with accompanying DNA damage
and genomic instability. Telomerase enzyme in normal HSCs preserves
long telomeres and promotes quiescence and a prolonged cellular
lifespan. Patients with autosomal dominant dyskeratosis congenita have
mutations in the genes for telomerase complexes, predisposing to
premature aging and enhanced marrow failure in the setting of
accelerated telomere shortening. One third of patients with acquired AA
also have short telomeres, likely due to a combination of genetic,
environmental, and epigenetic factors.

Autoreactive host lymphocytes can destroy normal hematopoiesis in
AA. Bone marrow stromal cells and cytokine levels in patients with
AA are normal.
The fact that AA also occurs in diseases of immune dysregulation and
after viral infections further suggests an immune-mediated
mechanism for the disease. One hypothesis is that drug or viral
antigens presented to the immune system trigger cytotoxicT-cell
responses that persist and destroy normal stem cells.
Only 1 in 100,000 patients develops severe AA as an idiosyncratic
drug reaction. Whether these individuals have a genetically
predisposed sensitivity to common exposures (e.g., nonsteroidal
anti-inflammatory drugs, sulfonamides, Epstein-Barr virus) is
unknown.

Clinical Presentation
The clinical onset of AA can be insidious or abrupt. Patients often
complain of symptoms related to their cytopenias: weakness, fatigue,
dyspnea, or palpitations resulting from anemia; gingival bleeding,
epistaxis, petechiae, or purpura caused by low platelet counts; or
recurrent bacterial infections caused by low or nonfunctioning
neutrophils.
Results of the physical examination are often normal except in
patients with congenital AA, who may have various abnormalities.

Diagnosis
Patients present with symptoms of bone marrow failure, usually anaemia
or bleeding, and less commonly, infections.
An FBC demonstrates pancytopenia, low reticulocytes and often
macrocytosis.
Bone marrow aspiration and trephine reveal hypocellularity. The severity
of aplastic anaemia is graded according to the Camitta criteria


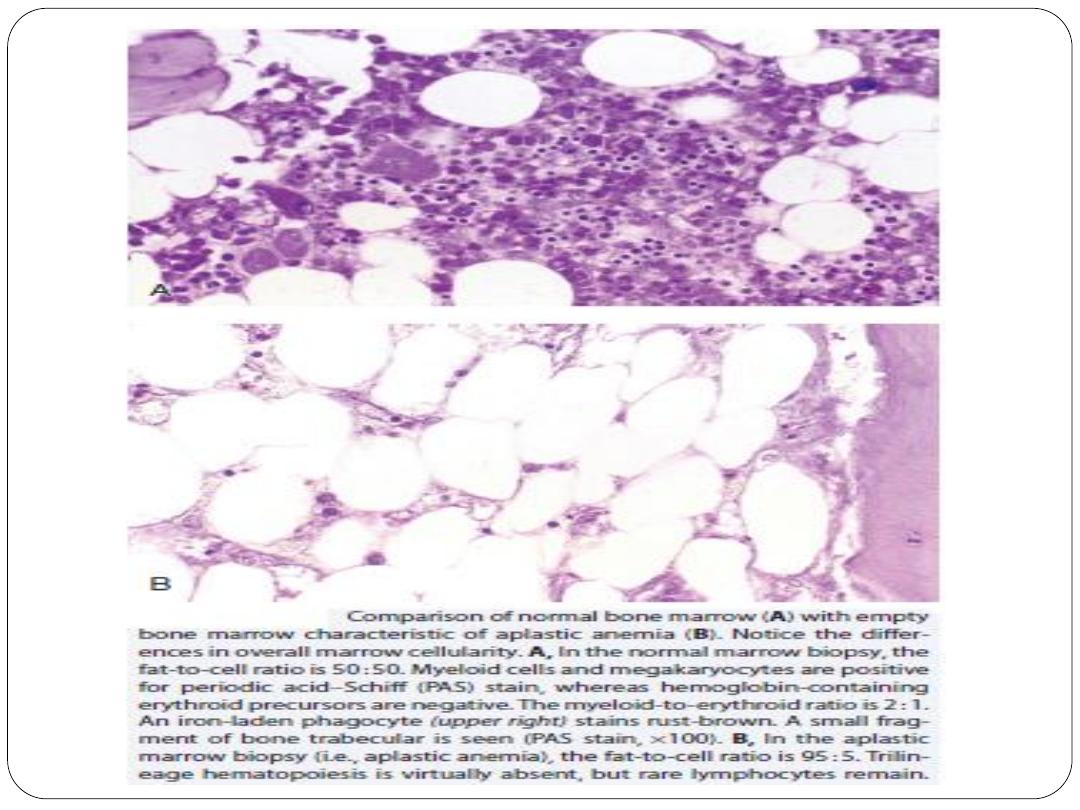
Diagnostic confirmation of AA requires bone marrow biopsy to
confirm hypocellularity and to rule out other marrow processes.
Normal bone marrow cellularity ranges from 30% to 50% up to
age 70 years and is less than 20% after 70 years of age.
In contrast, bone marrow cellularity in patients with AA usually
ranges from 5% to 15%, with increased fat accumulation and few
or no hematopoietic cells (primarily plasma cells and
lymphocytes)

In AA, hematopoietic progenitor and precursor
cells are morphologically normal but number
less than 1% of normal levels, and they are
markedly dysfunctional, with a decreased
ability to form differentiated progenitor cell
colonies in vitro.

Management
Treatment of AA is based on the severity of disease. Patients with mild
cytopenias can be monitored expectantly. However, patients with
severe AA based on peripheral blood cells counts (i.e., neutrophil count
<500/μL, platelet count <20,000/μL, anemia with corrected
reticulocyte count <1%, and marrow cellularity of 5% to 10%) have a
poor median survival of 2 to 6 months without treatment.
Because most of these patients die of overwhelming infections,
supportive care with broadspectrum antibiotics, antifungal agents, and
antiviral agents is warranted for those with advanced neutropenia. Red
blood cell and platelet transfusions can help patients who are
profoundly symptomatic, along with care given to patients eligible for
transplantation.

The curative treatment for patients under 35 years of age with
severe idiopathic aplastic anaemia is allogeneic HSCT if there is
an available sibling donor. Older patients (35–50) may be
candidates if they have no comorbidities. Those with a compatible
sibling donor should proceed to transplantation as soon as
possible; they have a 75–90% chance of long-term cure.
Although long-term survival is excellent for patients younger
than 30 years transplanted from a sibling donor (75% to 90%),
morbidity due to the transplant itself and the management of
long-term complications are continuing problems.
Outcomes for patients older than 40 years or patients without
an HLA-matched related donor are poor.

In older patients and those without a suitable donor,
immunosuppressive therapy (IST) with anti-thymocyte globulin
(ATG) and ciclosporin is the treatment of choice and gives 5-year
survival rates of 75%.
Unrelated donor allografts are considered for suitable patients
who fail immunosuppressive therapy .
Side effects of ATG include anaphylaxis and serum sickness as a
result of foreign antigens in the antisera, but these adverse effects
usually are self-limited.

Patients often relapse, and recurrence of disease may warrant
retreatment with ATG, androgens, and newer
immunosuppressive agents. Alemtuzumab, a humanized
monoclonal antibody directed against the CD52 protein found on
lymphocytes and which has efficacy in other autoimmune
diseases, has been as effective as rabbit ATG and cyclosporine in
relapsed and refractory severe AA.
Eltrombopag, an oral thrombopoietin receptor agonist (TPO)
mimetic drug that stimulates platelet production by binding to
MPL receptors on megakaryocytes, is an exciting agent for the
treatment of severe AA patients. Almost one half of patients
treated with eltrombopag exhibited clinically significant
responses in all three hematopoietic lineages, with normalization
of bone marrow cellularity and trilineage hematopoiesis.

Treatment of AA with traditional chemotherapy such as highdose
cyclophosphamide usually has proved too toxic.
Because endogenous cytokine production is usually high in
patients with AA, the routine use of growth factors such as G-CSF,
EPO, or stem cell factor typically is ineffective. However, in
patients with refractory disease, long-term administration of
combination cytokines may have some effect in sustaining blood
cell counts.

Non-transplanted patients may relapse and patients who survive
initial treatment of AA remain at increased risk for the
emergence of other primary hematologic disorders, such as
myelodysplasia, leukemia, and paroxysmal nocturnal
hemoglobinuria (PNH) for unknown reasons.
Patients with aplastic anaemia must be followed up long-term.

THANKS