Biochemistry
2nd stageDr.Lamees Majid Al-Janabi
Urea Cycle
Urea is the major disposal form of amino groups derived from amino acids, and accounts for about 90% of the nitrogen-containing components of urine. One nitrogen of the urea molecule is supplied by free NH3, and the other nitrogen by aspartate. The carbon and oxygen of urea are derived from CO2. Urea is produced by the liver, and then is transported in the blood to the kidneys for excretion in the urine.
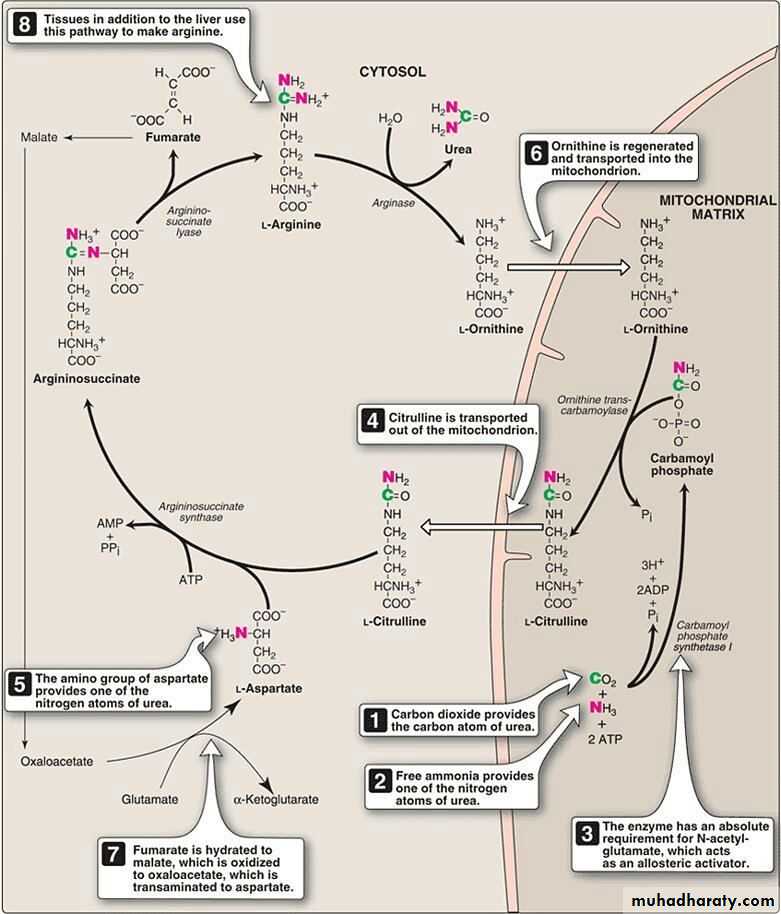
net reaction of urea cycle
carb. phosphate +aspartate+ 4 ATP →→→ urea + fumarate+ 2ADP + AMP+ PPiFate of urea:
Urea diffuses from the liver, and is transported in the blood to the kidneys, where it is filtered and excreted in the urine.
A portion of the urea diffuses from the blood into the intestine, and is cleaved to CO2 and NH3 by bacterial urease. This ammonia is partly lost in the feces, and is partly reabsorbed into the blood.
Urea is the principle end product of protein metabolism which is finally excreted in urine . normally the amount formed is dependent on protein intake . Urea cycle occur only in the liver because the enzyme Arginase not present in other tissue .
In patients with kidney failure, plasma urea levels are elevated, promoting a greater transfer of urea from blood into the gut. The intestinal action of urease on this urea becomes a clinically important source of ammonia, contributing to the hyperammonemia often seen in these patients.
The rate limiting step appear to be those catalyzed by the following enzyme :
-Carbamoyl phosphate synthetase
-Ornithine transcarbamoylase
-Arginase
Metabolism of Ammonia
Ammonia is produced by all tissues during the metabolism of a variety of compounds, and it is disposed of primarily by formation of urea in the liver. However, the level of ammonia in the blood must be kept very low, because even slightly elevated concentrations (hyperammonemia) are toxic to the central nervous system (CNS).
There must, therefore, be a metabolic mechanism by which nitrogen is moved from peripheral tissues to the liver for ultimate disposal as urea, while at the same time low levels of circulating ammonia must be maintained.
Sources of ammonia
From amino acids: Many tissues, but particularly the liver, form ammonia from amino acids by transdeamination—the linking of aminotransferase and glutamate dehydrogenase reactions .From glutamine: The kidneys form ammonia from glutamine by the actions of renal glutaminase and glutamate dehydrogenase. Most of this ammonia is excreted into the urine as NH4+, which provides an important mechanism for maintaining the body's acid-base balance. Ammonia is also obtained from the hydrolysis of glutamine by intestinal glutaminase. The intestinal mucosal cells obtain glutamine either from the blood or from digestion of dietary protein.
From bacterial action in the intestine: Ammonia is formed from urea by the action of bacterial urease in the lumen of the intestine. This ammonia is absorbed from the intestine by way of the portal vein and is almost quantitatively removed by the liver via conversion to urea.
From amines: Amines obtained from the diet, and monoamines that serve as hormones or neurotransmitters, give rise to ammonia by the action of amine oxidase .
From purines and pyrimidines: In the catabolism of purines and pyrimidines, amino groups attached to the rings are released as ammonia.
Hyperammonemia
The capacity of the hepatic urea cycle exceeds the normal rates of ammonia generation, and the levels of serum ammonia are normally low (5–50 µmol/L). However, when liver function is compromised, due either to genetic defects of the urea cycle, or liver disease, blood levels can rise above 1,000 µmol/L. Such hyperammonemia is a medical emergency, because ammonia has a direct neurotoxic effect on the CNS.For example, elevated concentrations of ammonia in the blood cause the symptoms of ammonia intoxication, which include tremors, slurring of speech, somnolence, vomiting, cerebral edema, and blurring of vision. At high concentrations, ammonia can cause coma and death. The two major types of hyperammonemia are:
Acquired hyperammonemia: Liver disease is a common cause of hyperammonemia in adults. It may be a result of an acute process, for example, viral hepatitis, ischemia, or hepatotoxins.
Hereditary hyperammonemia: Genetic deficiencies of each of the five enzymes of the urea cycle have been described, with an overall prevalence estimated to be 1:30,000 live births.
Glucogenic and Ketogenic Amino Acids
Amino acids can be classified as glucogenic, ketogenic, or both based on which of the seven intermediates are produced during their catabolism .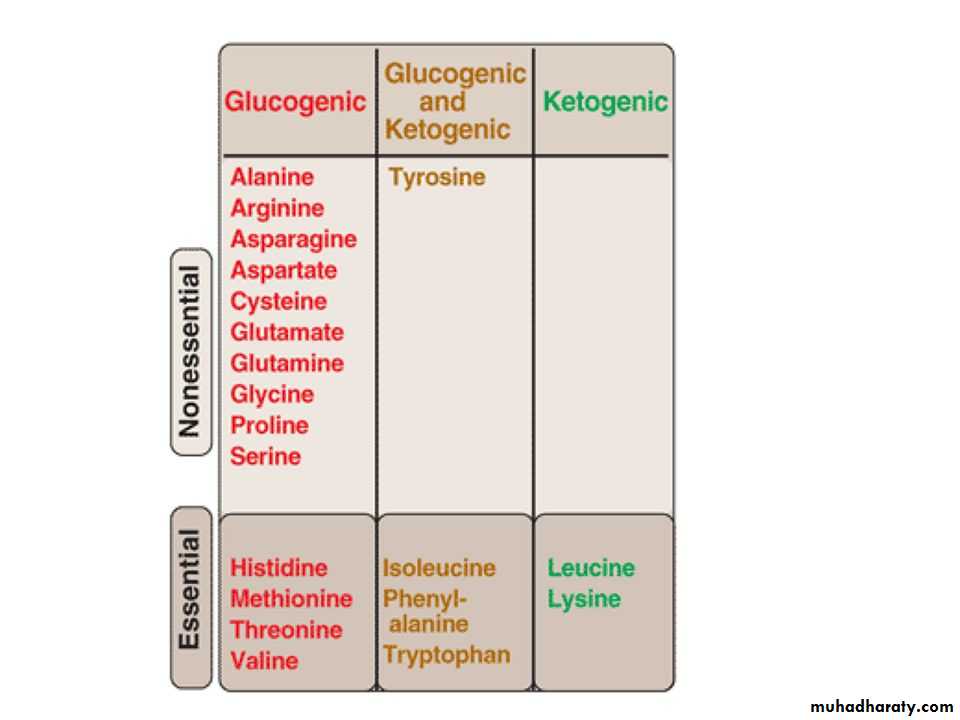
A. Glucogenic amino acids
Amino acids whose catabolism yields pyruvate or one of the intermediates of the citric acid cycle are termed glucogenic or glycogenic. These intermediates are substrates for gluconeogenesis and, therefore, can give rise to the net formation of glucose or glycogen in the liver and glycogen in the muscle.Ketogenic amino acids
Amino acids whose catabolism yields either acetoacetate or one of its precursors (acetyl CoA or acetoacetyl CoA) are termed ketogenic .Catabolism of the Carbon Skeletons of Amino Acids
The pathways by which amino acids are catabolized are conveniently organized according to which one (or more) of the seven intermediates listed above is produced from a particular amino acid.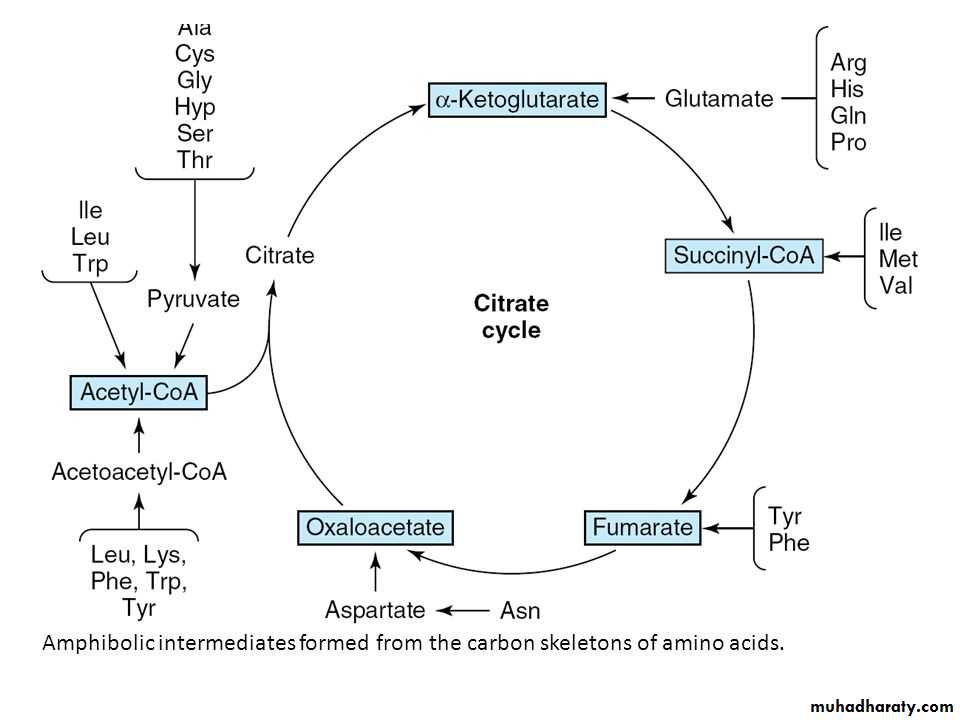
Amino acids that form succinyl CoA: methionine
Methionine is one of four amino acids that form succinyl CoA. This sulfur-containing amino acid deserves special attention because it is converted to S-adenosylmethionine (SAM), the major methyl-group donor in one-carbon metabolism . Methionine is also the source of homocysteine—a metabolite associated with atherosclerotic vascular disease.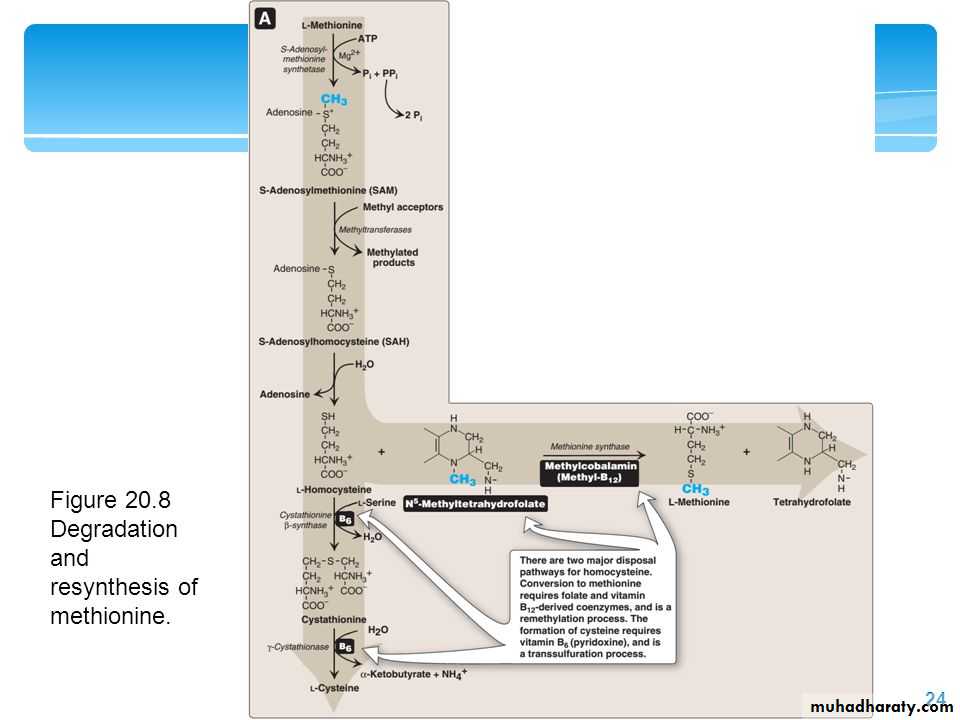
Relationship of homocysteine to vascular disease:
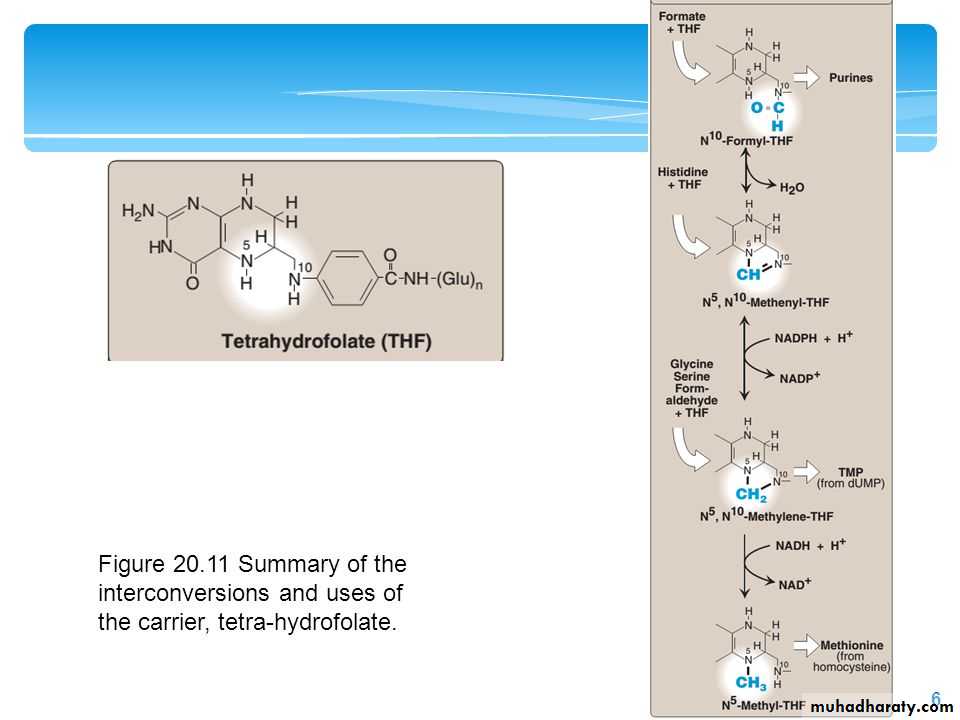
Elevations in plasma homocysteine levels promote oxidative damage, inflammation, and endothelial dysfunction, and are an independent risk factor for occlusive vascular disease . Mild elevations are seen in about 7% of the population.Role of Folic Acid in Amino Acid Metabolism
Folic acid: a carrier of one-carbon unitsThe active form of folic acid, tetrahydrofolic acid (THF), is produced from folate by dihydrofolate reductase in a two-step reaction requiring two moles of NADPH. The carbon unit carried by THF is bound to nitrogen N5 or N10, or to both N5 and N10. THF allows one-carbon compounds to be recognized and manipulated by biosynthetic enzymes.