

Autoregulation of renal blood flow and GFR
Despite changes in mean arterial blood pressure (from
80 to 180 mm Hg) , renal blood flow is kept at a relatively
constant level, a process known as
autoregulation Autoregulation is an intrinsic property
of the kidneys and is observed even in an isolated,
denervated, perfused kidney. GFR is also autoregulated
When the blood pressure is raised or lowered, vessels
upstream of the glomerulus (cortical radial arteries and
afferent arterioles) constrict or dilate, respectively,
maintaining relatively constant glomerular blood flow
and capillary pressure.

Below or above the autoregulatory range of
pressures, blood flow and GFR change appreciably
with arterial blood pressure. Renal autoregulation
minimizes the impact of changes in arterial blood
pressure on Na+ excretion. Without renal
autoregulation, increases in arterial blood
pressure would lead to dramatic increases in GFR
and potentially serious losses of NaCl and water
from the ECF.(Note: The autoregulatory
mechanisms of the kidney are not 100 % perfect).
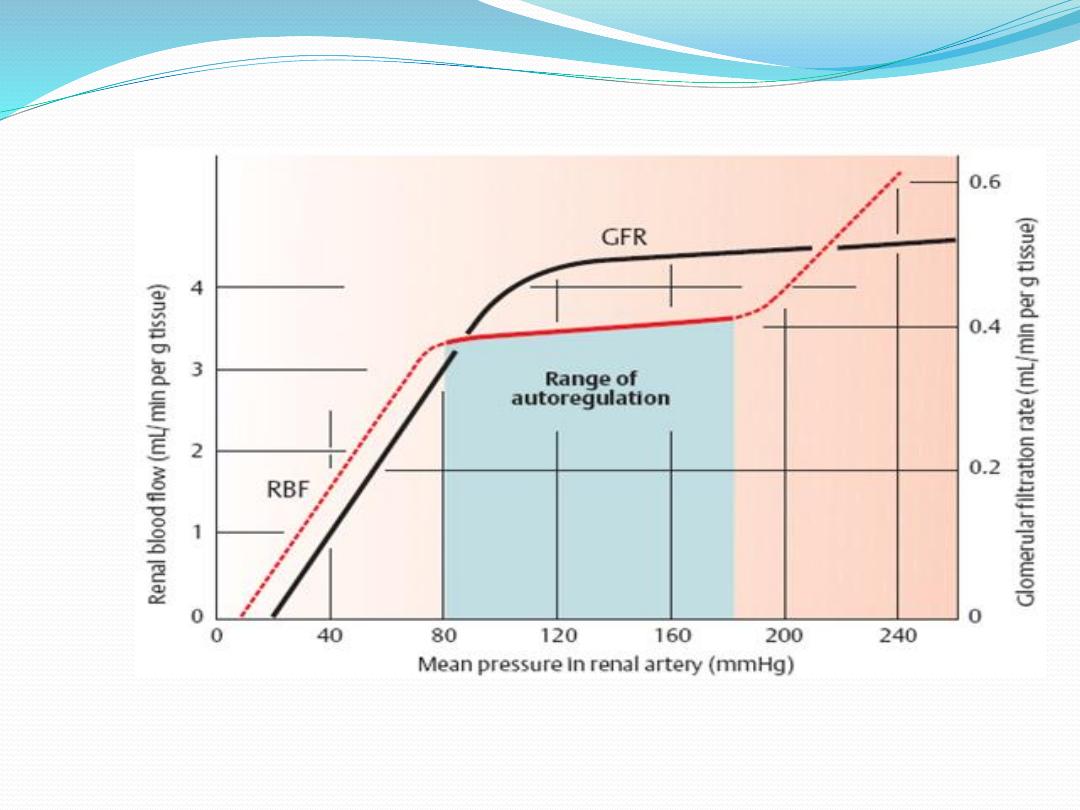
Autoregulation of renal blood flow and GFR

Two mechanisms account for renal autoregulation: the myogenic
mechanism and the tubuloglomerular feedback mechanism:
I.Tubuloglomerular feedback
This feedback mechanisms depend on juxtaglomerular complex which
consist of macula densa cells in the initial portion of the distal tubule
and juxtaglomerular cells in the walls of the afferent and efferent
arterioles. The macula densa cells sense changes in composition and
volume of fluid delivered to the distal tubule.
A decreased in blood pressure will decrease GFR this slows the fluid
flow rate in the loop of Henle, causing increased reabsorption of
sodium and chloride ions in the ascending loop of Henle, thereby
reducing the concentration of sodium chloride that reach the macula
densa cells. This decrease in sodium chlorideconcentration initiates a
signal from the macula densa that has two effects:

(1) it decreases resistance to blood flow in the afferent
arterioles, which raises glomerular hydrostatic pressure and
helps return GFR toward normal, and (2) it increases renin
release from the juxtaglomerular cells of the afferent and
efferent arterioles.Renin released from these cells then
functions as an enzyme to increase the formation of
angiotensin I, which is converted to angiotensin II. Finally,
the angiotensin II constricts the efferent arterioles, thereby
increasing glomerular hydrostatic pressure and returning
GFR toward normal.On the other hand increase in blood
pressure leads to increased solute delivery to the macula
densa .This produces an increase in the tubular fluid [NaCl]
at this site and increased NaCl reabsorption by macula
densa cells, leading to constriction of the nearby afferent
arteriole, thus, blood flow and GFR are lowered to a more
normal value.
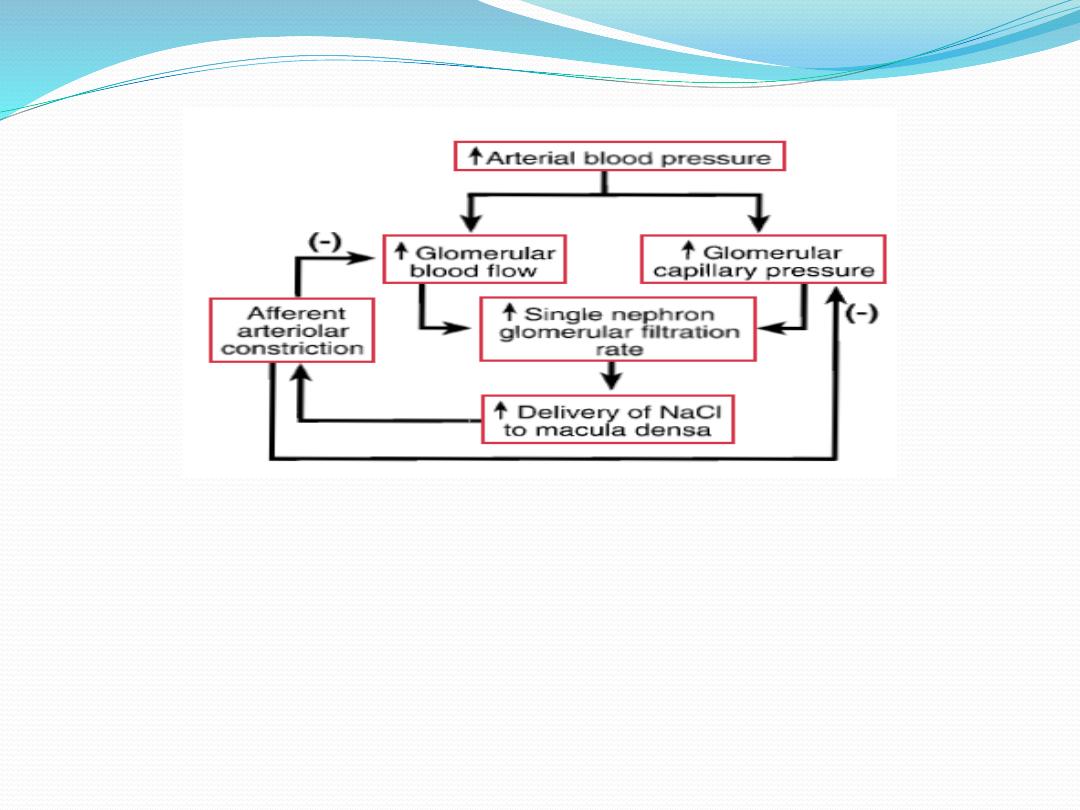
autoregulation feedback mechanism for increase in blood pressure.

II.Myogenic hypothesis
. The myogenic hypothesis states
that increased arterial pressure stretches the blood
vessels, which causes reflex contraction of smooth
muscle in the blood vessel walls and increased resistance
to blood flow . The mechanism of stretch-induced
contraction involves the opening of stretch-activated
calcium (Ca
2+
) channels in the smooth muscle cell
membranes. When these channels are open, more Ca
2+
enters vascular smooth muscle cells, leading to more
tension in the blood vessel wall. Decreased blood
pressure causes the opposite changes.

The filtration fraction
the filtration fraction is that fraction of the RPF that is filtered
across the glomerular capillaries. The filtration fraction is given by
the following equation:
The filtration fraction=GFR\RPF
The value for the filtration fraction is normally about r 20%. That
is, 20% of the RPF is filtered, and 80% is not filtered. The 80% of
RPF that is not filtered leaves the glomerular capillaries via the
efferent arterioles and becomes the peritubular capillary blood
flow.

Autocrine function of the kidney
Endothelins
The endothelins ET-1, ET-2 and ET-3 are a family of similar potent
vasoactive peptid es that also influence cell proliferation and
epithelial solute transport. They do not circulate but act locally. ETs
are produced by most types of cells in the kidney. The vascular
actions are mediated by two receptors,
1.ETA (specific for ET-1) mediating vasoconstriction and salt and
water retention and cause hypertension. Endothelins, mainly
through ETA receptors, can also alter cell proliferation and matrix
accumulation by increasing tissue inhibitor of metalloproteinase,
cytokines, fibronectin and collagen. These peptides also stimulate
the proliferation of a variety of renal cell types.
2. ETB (responsive to all ETs) causing vasodilatation, inhibit
sodium and water absorption by suppressing Na+/K+-ATPase and
Na+/H+ antiporter activity in the proximal tubule and antagonizing
the action of ADH and aldosterone in the collecting duct

Prostaglandins
Prostaglandins are unsaturated, oxygenated fatty
acids,derived from the enzymatic metabolism of
arachidonic acid, mainly by constitutively expressed cyclo-
oxygenase-1(COX-1) or inducible COX-2. COX-1 is highly
expressed in the collecting duct, while COX-2 expression is
restricted to the macula densa. Both COX isoforms convert
arachidonic acid to the same product, the bioactive but
unstable prostanoid precursor, prostaglandin H2 (PGH2).
PGH2 is converted to:
1. PGE2 (formed by PDE2 synthase in the collecting duct,
responsible for natriuretic and diuretic effects)
2. PGD2 (undetermined significance, produced in proximal
tubule)
3. prostacyclin (PGI2 ) (mainly synthesized in the
interstitial and vascular compartment
)

4. thromboxane A2
(vasoconstrictor, mainly synthesized in
glomerulus).
They all act through G-coupled transmembrane receptors.
Prostaglandins maintain renal blood flow and glomerular
filtration rate in the face of reductions induced by vasoconstrictor
stimuli such as angiotensin II, catecholamines and a-adrenergic
stimulation. In the presence of renal underperfusion, inhibition of
prostaglandin synthesis by non-steroidal anti –inflammatory
drugs results in a further reduction in GFR, which is sometimes
sufficiently severe as to cause acute renal failure. Renal
prostaglandins also have a natriuretic renal tubular effect and
antagonize the action of antidiuretic hormone. Renal
prostaglandins do not regulate salt and water excretion in normal
subjects, but in some circumstances, such as chronic renal failure,
prostagland ininduced vasodilatation is involved in maintaining
renal blood flow. Patients with chronic renal failure are thus
vulnerable to further deterioration in renal function on

exposure to non-steroidal anti-inflammatory
drugs, as are elderly patients in many of whom
renal function is compromised by renal vascular
disease and/ or the effect of ageing upon the
kidney. Moreover, in conditions such as volume
depletion, which are associated with high renin
release (facilitated by prostaglandins), inhibition
of prostaglandin synthesis may lead to
hyperkalaemia due to hyporeninaemic
hypoaldosteronism (since angiotensin II is the
mainstimulus for aldosterone).

Urodilatin: renal natriuretic peptide
A 32-amino-acid atrial natriuretic-like peptide
(ANP-like peptide), putatively synthesized by
connecting and collecting ucts in the kidney, has
been isolated from human urine. Its natriuretic
potency equals or exceeds that of atrial ANP by
increasing cGMP production in the collecting duct.
It is postulated that cardiac ANP is primarily a
regulator of the cardiovascular system through its
vascular effects and that renal natriuretic peptide
participates in the intrarenal regulation of sodium
and chloride transport.

Nitric oxide and the kidney
Nitric oxide, a molecular gas, is formed by the action of three
isoforms of nitric oxide synthase (NOS). All three enzymes,
neuronal (nNOS or NOS1), inducible (iNOS or NOS2) and
endothelial (eNOS or NOS3), which are cytochrome P450-like
proteins, facilitate the addition of the guanidine nitrogen of
the amino acid arginine to molecular oxygen, producing nitric
oxide and water. In general nNOS and eNOS are constitutively
active, producing low levels of nitric oxide dependent upon
intracellular calcium elevation. In contrast, the transcriptional
regulation of iNOS can be markedly induced, particularly by
inflammatory cytokines, resulting in extremely large amounts
of nitric oxide.

The most recognized cellular target of nitric oxide is soluble
guanylate cyclase. The stimulation of this enzyme enhances the
synthesis of cyclic GMP from GTP. All three isoforms, are expressed
in the kidney with eNOS in the vascular compartment, nNOS
mainly in the macula densa and inner medullary collecting duct
and iNOS in several tubule segments. Nitric oxide mediates the
following physiological actions in the kidney:
1.regulation of renal haemodynamics.
2.natriuresis by inhibiting Na+/K+-ATPaseandNa+/H+ antiporter
and antagonizing ADH.
3.modulation of tubuloglomerular feedback so that the
composition of tubular fluid delivered to the macula densa
changes the filtration rate of the associated
glomerulus.
