
1
Lec:3
Prof.Dr.Maha AlYasiri
HEMOSTASIS AND THROMBOSIS
Normal hemostasis
comprises a series of regulated processes that
culminate in the formation of a blood clot that limits bleeding from an
injured vessel.
The pathologic counterpart of hemostasis is thrombosis, the
formation of blood clot (thrombus) within non-traumatized, intact
vessels.
This discussion begins with normal hemostasis and its regulation, to
be followed by causes and consequences of thrombosis.
Normal Hemostasis
Hemostasis is a precisely orchestrated process involving
platelets, clotting factors
, and
endothelium
that occurs at the site of
vascular injury and culminates in the formation of a blood clot, which
serves to prevent or limit the extent of bleeding.
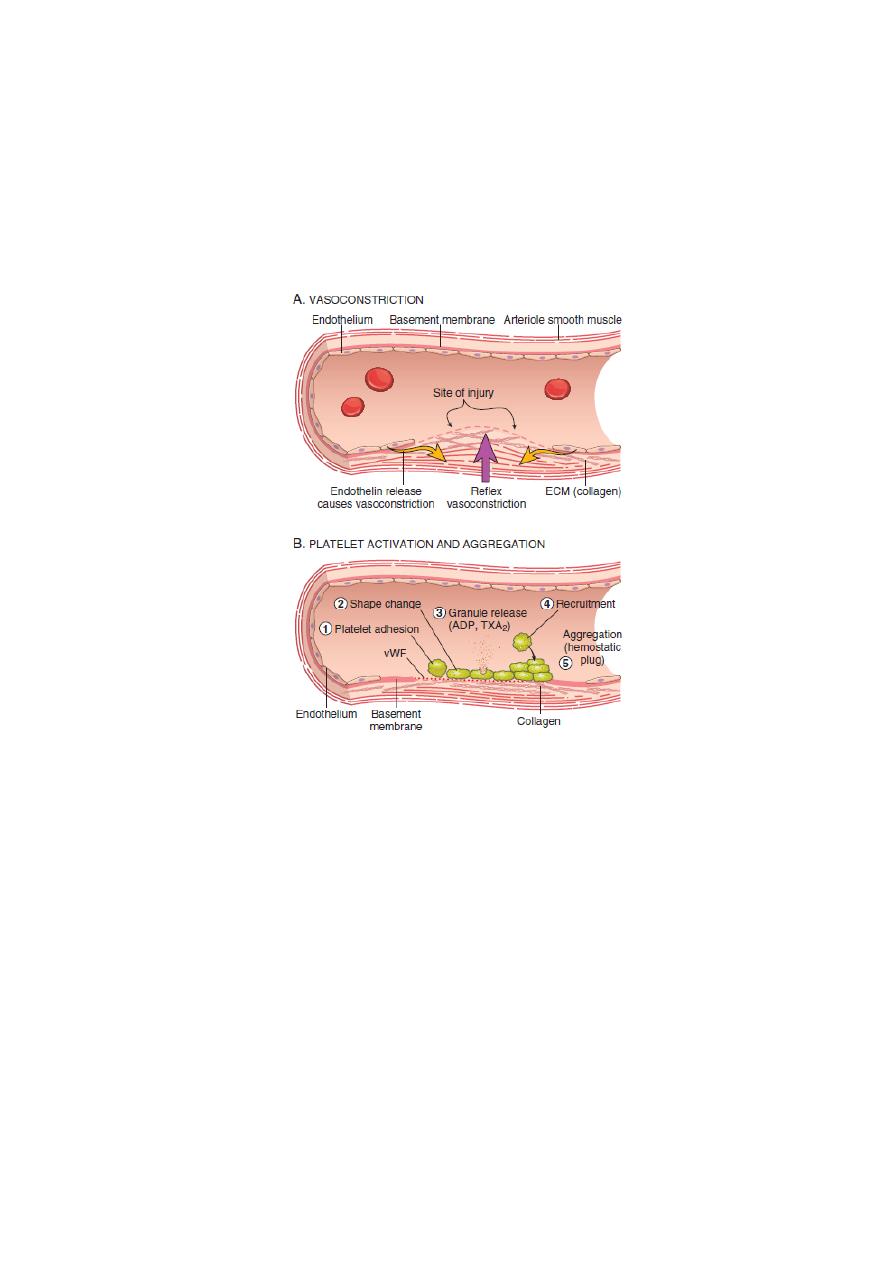
The general sequence of events leading to hemostasis at a site of
vascular injury:
(A) After vascular injury, local neurohumoral factors induce a
transient vasoconstriction.
(B)
Primary hemostasis: the formation of the platelet plug
. Platelets
bind via glycoprotein Ib (GpIb) receptors to von Willebrand factor
(VWF) on exposed ECM and are activated, undergoing a
shape
change
(from small rounded discs to flat plates with spiky
protrusions that markedly increased surface area) and
granule
release
. Within minutes the secreted products recruit additional
platelets, which undergo
aggregation
to form a
primary hemostatic
plug.
(C)
Secondary hemostasis: deposition of fibrin.
Vascular injury
exposes tissue factor at the site of injury. Tissue factor is a
glycoprotein that is normally expressed by subendothelial cells in
the vessel wall, such as smooth muscle cells and fibroblasts. Tissue
factor binds and activates factor VII, setting motion a cascade of
reactions that culiminates in
thrombin generation
. Thrombin cleaves
circulating
fibrinogen into insoluble fibrin
, creating a fibrin
meshwork, and also is a
potent activator of platelets
, leading to
additional platelet aggregation at the site of injury. This sequence,
referred to as secondary hemostasis, consolidates the initial platelet
plug
definitive secondary hemostatic plug.
2
(D)
Clot stabilization and resorption.
Polymerized fibrin and platelet
aggregates undergo contraction to form a solid, permanent plug that
prevents further hemorrhage. At this stage, counterregulatory
mechanisms (e.g., tissue plasminogen activator, t-PA made by
endothelial cells) are set into motion that limit clotting to the site of
injury and eventually lead to clot resorption and tissue repair.
3
Fig. Normal hemostasis.
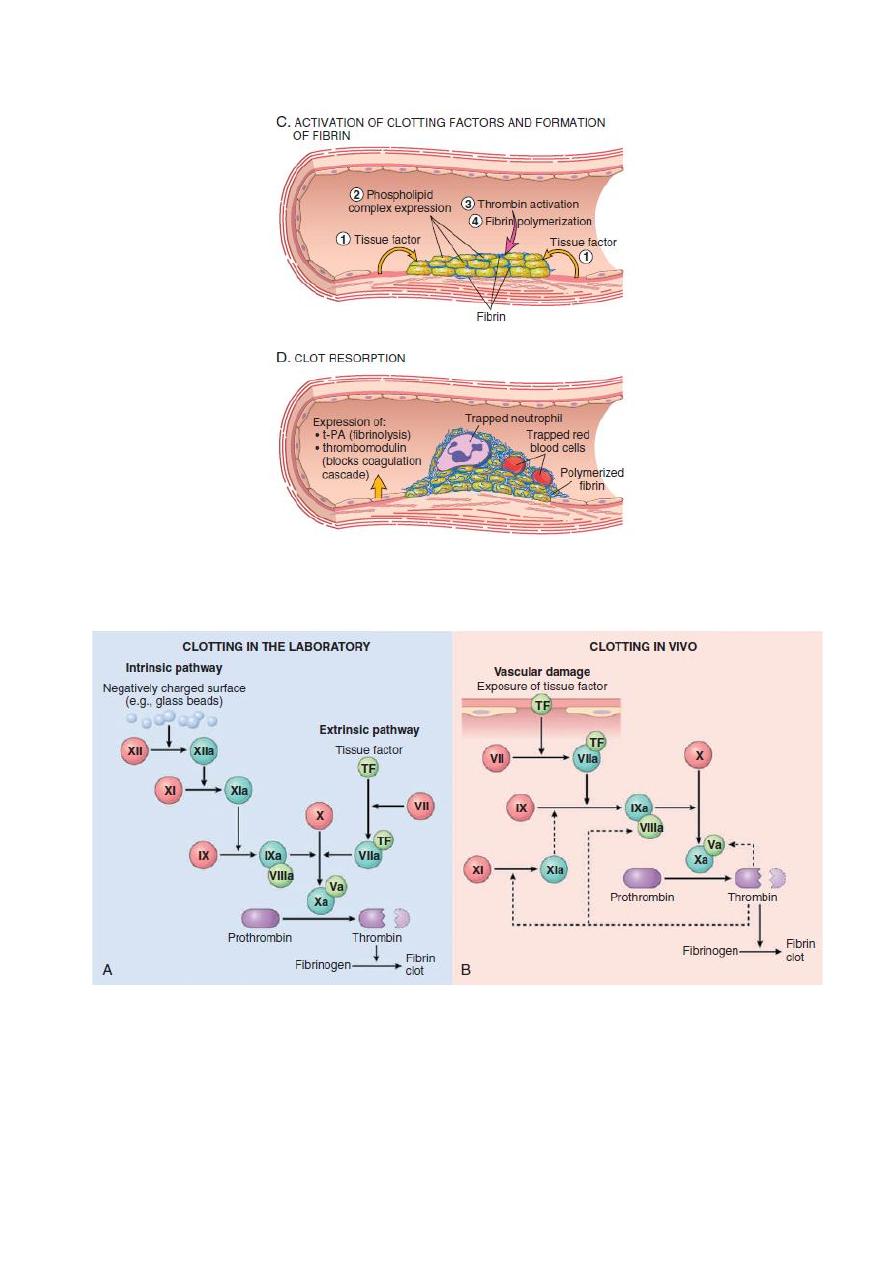
Fig. The coagulation cascade in the laboratory and in vivo. (A) Clotting is initiated
in the laboratory by adding phospholipids, calcium, and either a negative-charged
substance such as glass beads (intrinsic pathway) or a source of tissue factor
(extrinsic pathway). (B) In vivo, tissue factor is the major initiator of coagulation,
which is amplified by feedback loops involving thrombin (dotted lines). The red
polypeptides are inactive factors, the dark green polypeptides are active factors,
whereas the light green polypeptides correspond to cofactors.
4
Thrombosis
The pathologic counterpart of hemostasis is thrombosis, the
formation of blood clot (thrombus) within non-traumatized, intact
vessels.
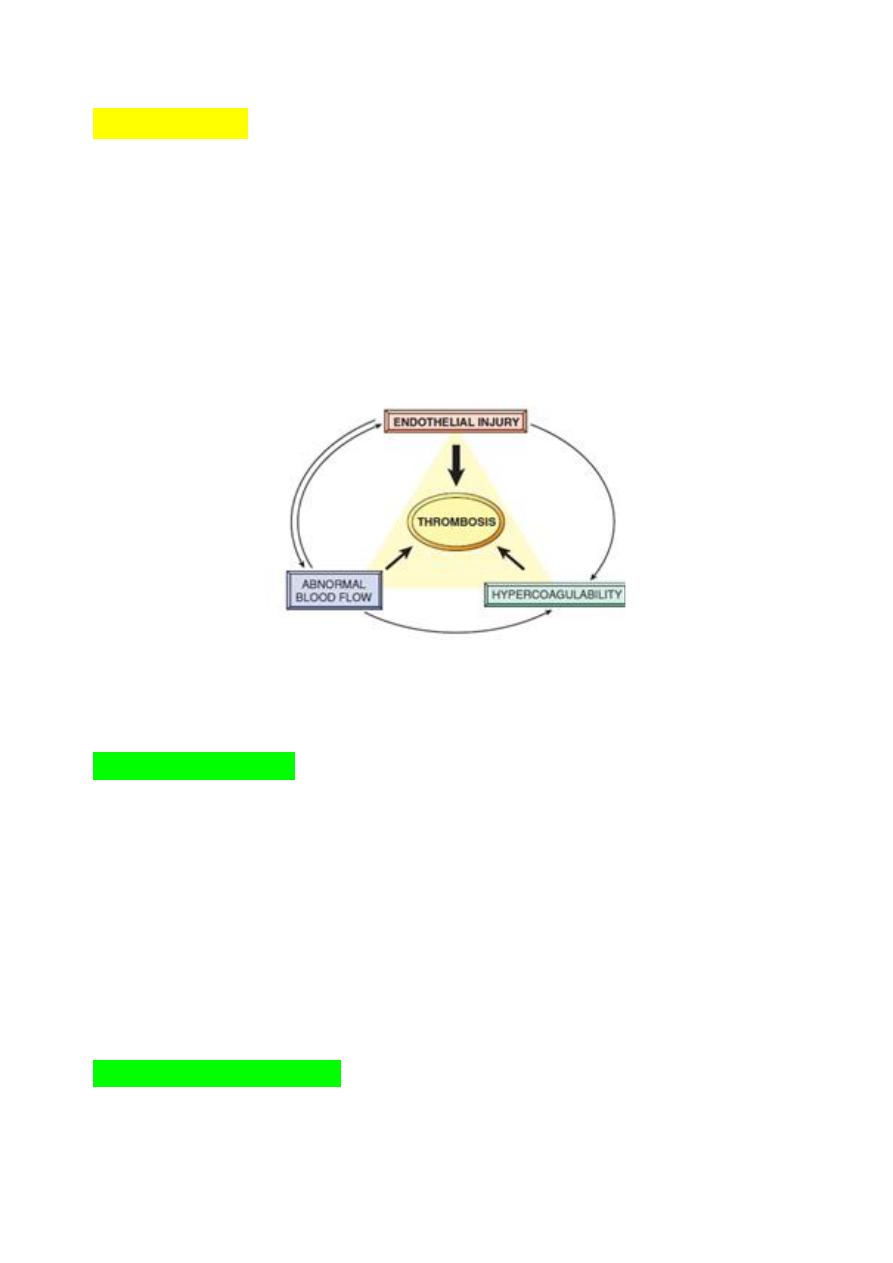
The primary abnormalities that lead to intravascular thrombosis are:
(1) endothelial injury,
(2) stasis or turbulent blood flow, and
(3) hypercoagulability of the blood
(the so-called “Virchow triad”).
Thrombosis underlies the most serious and common forms of
cardiovascular disease.
Fig. Virchow’s triad in thrombosis. Endothelial integrity is the most important
factor. Abnormalities of procoagulants or anti-coagulants can tip the balance in
favor of thrombosis. Abnormal blood flow (stasis or turbulence) can lead to
hypercoagulability directly and also indirectly through endothelial dysfunction.
Endothelial injury
• Endothelial injury leading to platelet activation.
• underlies thrombus formation in the heart and the arterial
circulation, where the high rates of blood flow impede clot formation
by preventing platelet adhesion or diluting coagulation factors.
• severe endothelial injury may trigger thrombosis by exposing VWF
and tissue factor.
• Most common examples are:
#Endocardial injury during myocardial infarction.
#Injury over ulcerated plaque in severely atherosclerotic arteries.
#vasculitis.
Abnormal Blood Flow
Turbulence
(chaotic blood flow) contributes to arterial and
cardiac thrombosis by causing endothelial injury or dysfunction,

5
as well as by forming countercurrents and local pockets of
stasis.
Stasis
is a major factor in the development of venous thrombi.
Under conditions of normal laminar blood flow, platelets (and
other blood cells) are found mainly in the center of the vessel
lumen, separated from the endothelium by a slower-moving layer
of plasma.
By contrast, stasis and turbulence have the following deleterious
effects:
• Both promote endothelial cell activation and enhanced
procoagulant activity.
• Stasis allows platelets and leukocytes to come into contact with
the endothelium when the flow is sluggish.
• Stasis also slows the washout of activated clotting factors and
impedes the inflow of clotting factor inhibitors.
Turbulent and static blood flow contributes to
thrombosis in a number of clinical settings.
•
Ulcerated
atherosclerotic
plaques
not
only
expose
subendothelial ECM but also cause turbulence.
•
Abnormal aortic and arterial dilations called aneurysms
create
local stasis and consequently are fertile sites for thrombosis .
•
Acute myocardial infarction
results in focally noncontractile
myocardium. Ventricular remodeling after more remote
infarction can lead to aneurysm formation. In both cases,
cardiac mural thrombi are more easily formed because of the
local blood stasis.
•
Mitral valve stenosis
(e.g., after rheumatic heart disease)
results in left atrial dilation. In conjunction with atrial
fibrillation, a dilated atrium also produces stasis and is a prime
location for the development of thrombi.
•
Hyperviscosity syndromes
(such as polycythemia vera,)
increase resistance to flow and cause small vessel stasis;
•
sickle cell anemia
the deformed red cells cause vascular
occlusions, and the resultant stasis also predisposes to
thrombosis.
Hypercoagulability
Hypercoagulability refers to an abnormally high tendency of the
blood to clot, and is typically caused by alterations in coagulation
factors.
It contributes infrequently to arterial or intracardiac thrombosis
but is an important underlying risk factor for venous thrombosis.

6
The alterations of the coagulation pathways that predispose
affected persons to thrombosis can be divided into primary
(genetic) and secondary (acquired) disorders.
Primary (Genetic)
Common (>1% of the Population)
• Factor V mutation
• Prothrombin mutation
• Increased levels of factor VIII, IX, or XI or fibrinogen
Rare
• Anti-thrombin III deficiency
• Protein C deficiency
• Protein S deficiency
Secondary (Acquired)
• Prolonged bed rest or immobilization
• Myocardial infarction
• Atrial fibrillation
• Tissue injury (surgery, fracture, burn)
• Cancer
• Prosthetic cardiac valves
• Disseminated intravascular coagulation
• Heparin-induced thrombocytopenia
• Anti-phospholipid antibody syndrome
MORPHOLOGY
Thrombi can develop anywhere in the cardiovascular system.
Arterial or cardiac thrombi
typically arise at sites of endothelial injury
or turbulence;
venous thrombi
characteristically occur at sites of stasis.
Thrombi are
focally attached to the underlying vascular surface
and
tend to
propagate toward the heart;
thus,
arterial thrombi
grow in a
retrograde direction from the point of attachment, whereas
venous
thrombi
extend in the direction of blood flow.
The propagating portion of a thrombus tends to be poorly attached
and therefore prone to fragmentation and migration through the blood
as an
embolus.
Thrombi can have grossly (and microscopically) apparent laminations
called
lines of Zahn
; these represent pale platelet and fibrin layers
alternating with darker red cell–rich layers. Such lines are significant
in that they are only found in thrombi that form in flowing blood; their
presence can therefore usually distinguish antemortem thrombosis
from the bland nonlaminated clots that form in the postmortem state.

7
Although thrombi formed in the “low-flow” venous system
superficially resemble postmortem clots, careful evaluation generally
shows ill-defined laminations.
Thrombi occurring in heart chambers or in the aortic lumen are
designated as
mural thrombi.
Arterial thrombi
are frequently occlusive. They are typically rich in
platelets, as the processes underlying their development (e.g.,
endothelial injury) lead to platelet activation. Although usually
superimposed on a ruptured atherosclerotic plaque, other vascular
injuries (vasculitis, trauma) can also be underlying causes.
Venous thrombi
(phlebothrombosis) are almost invariably occlusive;
they frequently propagate some distance toward the heart, forming a
long cast within the vessel lumen that is prone to give rise to emboli.
Because these thrombi form in the sluggish venous circulation, they
tend to contain more enmeshed red cells, leading to the moniker red,
or stasis, thrombi. The veins of the lower extremities are most
commonly affected (90% of venous thromboses
At autopsy, postmortem clots can sometimes be mistake for venous
thrombi. However, the former are:
• gelatinous
• and because of red cell settling they have a dark red dependent
portion and a yellow “chicken fat” upper portion;
• they also are usually not attached to the underlying vessel wall.
By contrast, red thrombi typically are:
• firm,
• focally attached to vessel walls,
• and the contain gray strands of deposited fibrin.
Thrombi on heart valves are called vegetations.

8
Fig. Mural thrombi. (A) Thrombus in the left and right ventricular
apices, overlying white fibrous scar. (B) Laminated thrombus in a
dilated abdominal aortic aneurysm. Numerous friable mural thrombi
are also superimposed on advanced atherosclerotic lesions of the
more proximal aorta
(left side of photograph).