Haematology
Dr Khudhair Abass AliCollege of Medicine – Baghdad University
Objectives L7
Hematological malignancies: Acute Leukemias:Lymphoblastic(ALL),Myeloblastic(AML)
Chronic Leukemias:
Myeloblastic(CML),Lymphoblastic(CLL)Haematological Malignancies
Corruption of the processes controlling proliferation & apoptosis of bl. Cells.If mature cells involved → Chronic Leukemia or Low grade Lymphoma
Primitive cells involved → Acute Leukemia or High grade Lymphoma
Leukemias : 100/ 100 000 /y .Acute Leukemia ˂ 50 % M ˃ F ,
Acute: All ages , ALL ↑ in children, AML ↑ in Old.
Risk Factors : 1. Ionizing radiation
2. Cytotoxic drugs
3.Retroviruses.
4.Genetics
5.Immunological.
Lecture (7)
Acute Leukemia : ALL & AML
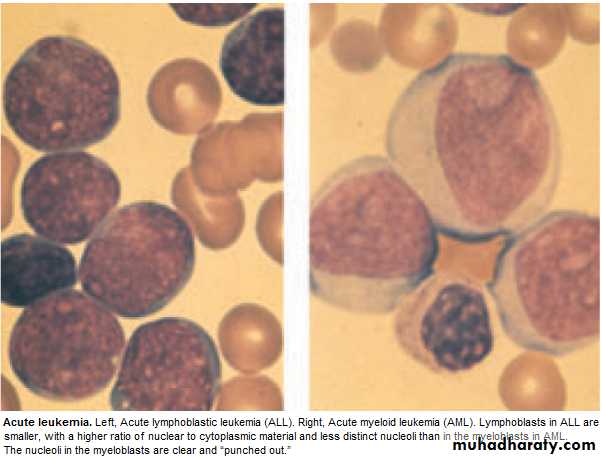
Failure of cell maturation, accumulation Of useless immature cells in B.M. on theexpense of normal haemopoeitic elements, with spilling of immature cells into Bl.
Clinical Features are: marrow failure ( Anemia, Bleeding, or Infection)
Investigations :
CBP : Anemia ,WBC : ↓ 1 X 10 9/ L --- ↑ 500 X 10 9/ L (Majority ˂ 100 X 10 9/L. Platelets : usually very low.
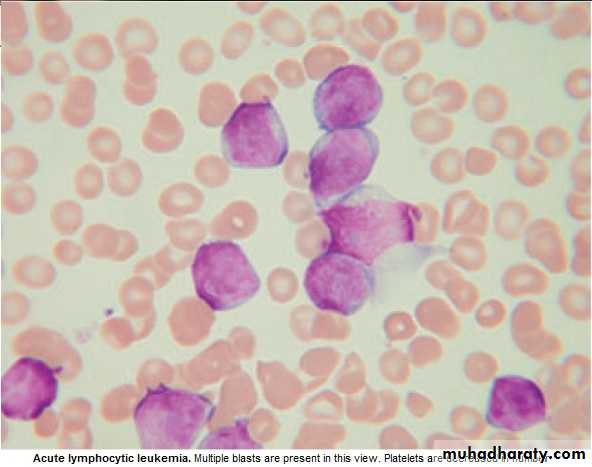
BLAST in Peripheral Blood .
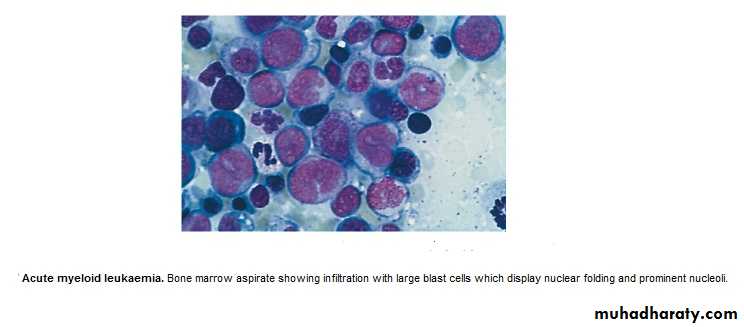
BM : Confirm the diagnosis, replacement of normal elements by leukemic
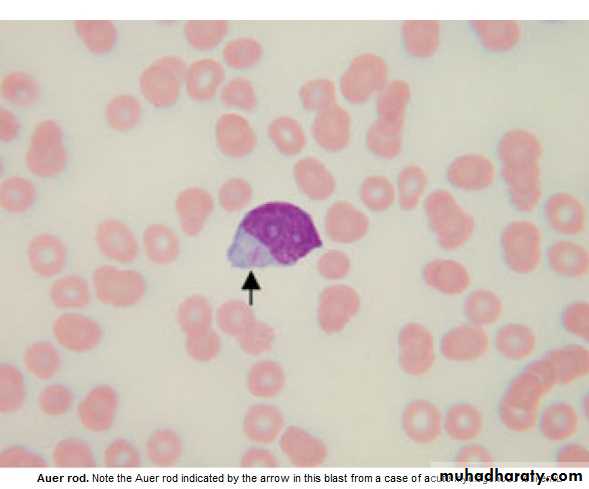
blast cells ( ˃ 20 % of the cells ),presence of Auer rods in the cytoplasm
of the blast cells = AML.
Management :
1. Decide whether to give specific treatment or not,(It is aggressive with ↑S.E.)
which may not be appropriate for elderly or patients with other serious dis.→ supportive treatment.
Specific Treatment :
Preparation for specific treat. :1.Identify & treat infection
2.Correct Anemia :Red cell Concentrate.
3.Thrombocytopenic bleeding ← Platelets'.
4.Hicman line (if possible)
5.Explain Therapeutic regimen to pt. & obtain consent.
-Aim of Treat. = destroy leukemic cells without destroying the normal stem cells
→Repopulation of haemopoeitic Ts.
Remission induction :Bulk of tumor is destroyed by combination chemotherapy
→Severe bone marrow hypoplasia (require intensive support.)
Remission consolidation :If Rem. Achieved → Residual dis. attacked by No. of courses of chemo.→ marrow hypoplasia (BMT may be included at this stage)
Rrmission maintenance :If still in Rem.(ALL) → Repeated cycles of Chemo. up to 3yr.
+ CNS prophylaxis = Intathecal (IT) Methotrexate + cranial radiation.
Failure of Rem. in induction. →Alternative combination of drugs.(poor prognosis)
Relapse during treat. Or after → poor prognosis.
Drugs commonly used in treat. Of Acute Leukaemia
phase ALL AML
Induction Vivcristine (iv) Daunorubicine(iv)
Prednisolone(po) Cytarabine(iv)
L-asparaginase(im) Etoposide(iv&po)
Daunorubicine(iv)
Methotrexate(IT)
Consolidation Daunorubicine(iv) Cytarabine(iv)
Cytarabine(iv) Amsacrine(iv)
Etoposide(iv) Mitoxantrone(iv)
Methotrexate(iv)
Maintenance Prednisolone(po)
Vincristine(iv)
Mercaotopurine(po)
Methotrexate(po)
Supportive treatment:
Anemia : Red Cell Concentrate Transfusion.Bleeding :Thrombocytopenic bleeding require platelets.Prphylactic plat.transf.to
maintain plat.˃ 10 X 10 9/L. Treat any Coagulation abnormalities.
Infection : Fever ˃ 38oC , ˃ 1h in Neutropenic pt. indicate possible septicemia. Start
Broad spectrum antibiotic(Gentamycin + piperacillin± Vancomycin),to continue for at least 3ds after the fever resolved.
Flucanazole for oral & pharyngeal moniliasis.
Amphotericin iv for systemic fungal inf.(candida or aspergillosis) for 3wks.
Herpes simplex and zoster : antiviral(aciclovir).
Isolation is debatable (psychological).
Metabolic problems :Monitor fluid, hepatic, renal function,anorexia & difficult drinking
→ iv fluid & electrolytes. Cellular breakdown during induction(tumor lyses'
syndrome) → ↑K, ↑Uric acid, ↑phosphate,& ↑Ca. → RF ?
Allopurinol + iv Hydration .
Psychological : Keep pt. informed ,answer question, allay fear as possible.
Prognosis : Median survival without treat.is 5wks….Few Ms with supportive treatment.
80% ˂ 60y age achieve remission with specific treat.
Survival depends on risk factors (chromosomal analysis)
5y survival for AML 21%-76%, ALL 20-37% .
Chronic Myeloid Leukemia ( CML )
* Myeloproliferative stem cell disorder resulting in prolif. Of all hematopoietic
Lineage but predominantly in the granulocytes series. Chiefly 30-80y, 20% of all
leukemia. * 95% have Philadelphia (ph ch) = shortened ch. 22 resulting from translocation
of material with ch 9.
* BCR(breakpoint cluster region on chr.22 + Fragment from chr. 9 carries ABL oncogen → BCR ABL Gene codes for protein with Tyrosine Kinas activity→ play a role in the dis. As an oncogen.
CML has 3 phases:
1.Chronic phase:. The dis. is responsive to treat., lasting 3-5y. With the introduction of imatinib → ˃ 5Y.
2.Accelerated phase :(not always seen), dis. control more difficult.
3.Blast crises:dis transforms into acute leukemia (AML%70 ,ALL30%),Refractory to
treat., It is the major cause of death. Prior to imatinib therapy, 10% CML→ AML/y, now only 0.4-2.5% after up to 5y treatment with imatinib.
ph. Ch. –ve CML: 0ld pt., ↑Male, ↓Plat. ↑ Monocyte,respond poorly to treat.
Median survival 1y.

Clinically: 25% - asymptomatic at diagnosis.
Tiredness 37% Anorexia 12%SOB 21% Abdom. Fullness 10%
Abdom. Pain 21% Bruising 7 %
Lethargy 13% Vague ill health 7%
*Splenomegally 90% (10% massive) ± friction rub(splenic infarction)
*Hepatomegally 50%
*LN Unusual.
Investigations:1.FBC: An.(normochromic normocytic).WBC 10-600X10 9/L(full range
of granulocytes precursors from myeloblast to mature neutrophils,
myeloblast ˂ 10%, ↑Eosinoph .&Basophils.
Platelets: ↑ in ⅓ (up to 2000x109/L . Nucleated RBC are common.
Accelerated phase: ↑ % of more primitive cells.
Blast transformation: Dramatic ↑ Blast in circul., ↓plat.
Basophils ↑ with dis. Progress.
2. Bone marrow: to confirm diagnosis &phase of the dis.(morphology & chromosome analysis .
3. Bl. LDH & Uric acid ↑ ( ↑ cell breakdown)
Management:
Chronic phase:1.Imatinib inhibit BCR ABL Tyrosine Kinase(TK) activity & reduce
. the uncontrolled prolif.of WBC.. It is Ist line therapy in chronic phase
complete cytogenic response (disappearance of ph chr.) in 76% at 18 Ms of therapy.
Failure of response or progress on imatinib → 2nd generation TK Inh. e.g.
Dasatinib, or Nilotinib : 2. Allogenic BMT
3. Hyroxycarbamide (Hydroxyurea):still used for initial control of the dis.or
palliative treat.(no effect on ph chr. & on onset of blast transformation).
4. Interferon-ἀ : was Ist line of treat.± Ara-C: control CML in 70% of cases.
Accelerated phase: Pt. presented with this phase ,imatinib is indicated if not already
received. Hydroxyurea is also effective. Low dose cytarabine can be trie Blast Transform.: ALL response better than AML.( + supportive treat.)
Pts. Progressing to advanced phase on imatinib may respond to 2nd genera. TK inh. or BMT.
Chronic Lymphocytic Leukemia (CLL)
* Most common leukemia(30%).M:F is 2:1, median age at presentation 65-70y.* B lymphocytes fail to transform & produce Abs →Increasing mass of immuno-
incompetent cells → ↓immune function & normal BM haematopoeisis.
Clinically:
*insidious onset. 70% incidental diagnosis (routine FBC).
*Anemia, Infection, Painless Lymphadenopathy ± splenomegally & systemic symptoms e.g.Night sweat ,or wt. loss.
Investigations:
1.FBC: Mature lymphocytes ˃ 5x 10 9/L. characteristic morphology & markers, CD19 & CD23.
2.↑ Reticulocyte count & +ve coombs test.= haemolytic An.(may occur)
3.S. Igs: to assess the degree of immunosuppression(common & progressive).
4.B.M. exam. Is not essential for diagnosis, but helpful for prognosis(diffuse involve.= poor prognosis),& to monitor response to therapy.
Main prognostic factor is stage of the dis.,CD38 ,Mutation of IgVH Genes suggest poor prognosis.
Staging of CLL:
Stage A: 60%,No anemia, Normal Plat. Count, ˂ 3 areas of LN enlargement.
Stage B: 30%,No anemia, Normal Plat., 3 or more areas of LN enlarg.
Stage C : 10%,Anaemia & or ↓ Plat., regardless of the No. of areas of LN enlarg .
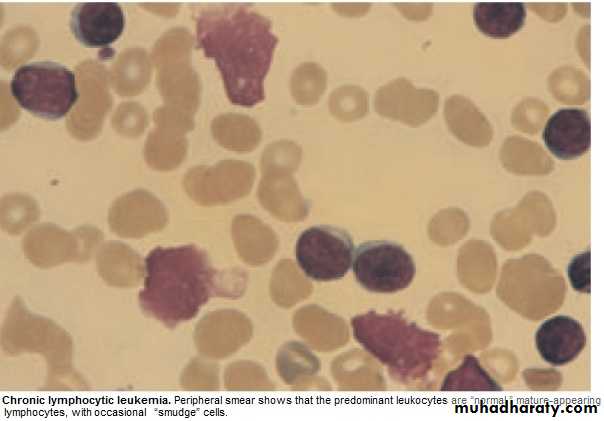
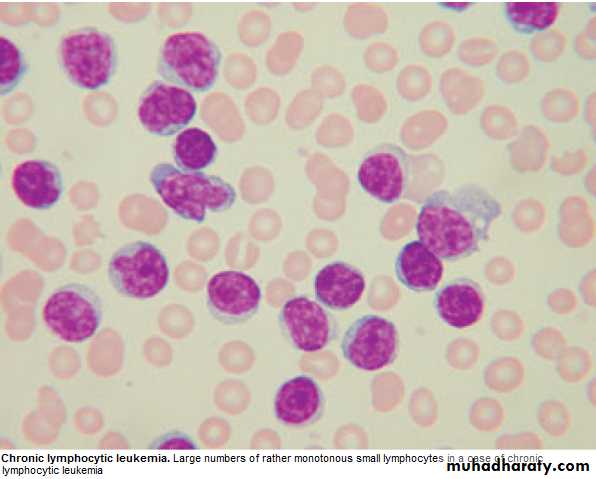
Management:
* No specific treat. For most stage A, unless progression, life expectancy is normal inmost patients.
*Treatment: Indications: 1. BM failure. 2. Massive or progressive LN PATHY or Splenomegally.
3. Systemic symptoms e.g. wt. loss or night sweat.
4. Rapidly ↑ lymphocyte count.
5. Autoimmune haemolytic An.,or ↓ Plat.
Stage B & C :Chlorambucil
Recently Fludarabine + Cyclophosphamide → ↑ rem.rates & dis. Free survival ( ↑ risk of infection )
B.M. Failure or autoimmune cytopenias → corticosteroid.
Supportive care: Symptomatic, Anemia or thrombocytopenia → Transfusion.
Treat. Of infect., Ig for hypogammaglobulinaemia.
Radiotherapy: LN causing discomfort or obstruction.& for symptomatic splenomegaly Splenectomy : may be , to improve low Bl. Count. due to autoimmune destruction or due to hypersplenism & can relieve massive splenomegally.
* Prognosis: Overall survival is 6y., stageC 2-3y, ,50% die from infection.
Rarely → Aggressive high grade Lymphoma(Richters syndrome).
Prolymphocytic Leukemia
* Variant of CLL, mainly in Male ˃ 60y. 25% T cell variety* Massive splenomegaly, + Little Lymphadenopathy.
* WBC often ˃ 400 x 10 9/ L (Characteristic cell is large lymphocyte with prominent nucleolus.
* Treat. : Is generally unsuccessful & prognosis very poor.
Leukapharesis, Splenectomy, & chemo. May be tried.
Hairy cell Leukemia :
* Rare,chronic lymphoproliferative B cell disorder, M 6:F 1 , median age 50y.* General ill health, & recurrent infection.
* Splenomegaly 90%, LN unusual.
* Lab.: Severe Neutropenia, Monocytosis,& characteristic HAIRY CELL in Bl. & B.M..
→ CD25 & CD103.
* Treat.: Cladribine & Deoxycoformycin.
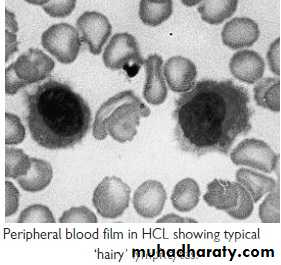
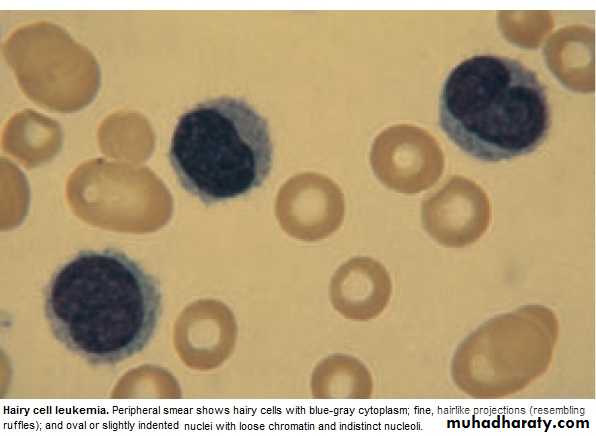
Summary
Acute leukemias: ALL & AMLFailure of maturation.
Lab: > 20% Blast.in BM.
R: Supportive or aggressive
CML:↑profil. Of all haemopo.cells.
90% +ve ph chr.
3 phases: Acute, Accelerated, Blast.
R:Imatinib
CLL: B Lymphocytes fail to transform & produce Ab.
3 stages.
R:Fludarabine