Inherited glomerular diseases & Cystic kidney diseases
Dr. Mohammed Hannon Al Sodani C.A.B.M., F.I.C.M.S.For 5th year medical students 2012-2013
College of Medicine , University of BaghdadMarch,24th,2016
Inherited glomerular diseases
Uncommon diseases may affect the glomerulus in childhood,
-The most important one affecting adults is
Alport’s syndrome
- 5% of ESRD in childhood or adolescence.
- Most common X-linked recessive 85%,
from a mutation or deletion of the COL4A5 gene , encodes
type IV collagen
- less common autosomal recessive disease Mutations in COL4A3
or COL4A4 genes
- The accumulation of abnormal collagen results in a progressive
degeneration of the GBM .
Inherited glomerular diseases Alport’s syndrome
Affected patients with
- haematuria,
- proteinuria (less than 1–2 g/day),
- progressive to ESRD in their late teens or twenties.
Female carriers usually have haematuria but rarely develope significant renal disease.
- Cochlea involvement ,
high-frequency sensorineural deafness
- Ocular involvement
15% anterior lenticonus and macular and perimacular
retinal flecks
Alport’s syndrome
No specific treatment is available,early , ACE inhibitors may attenuate proteinuria.
experimental ;- that mesenchymal stem cells can transdifferentiate into podocytes and repair basement abnormalities and slow the progression.
but patients with Alport’s syndrome are good candidates for RRT, as they are young and usually otherwise healthy.
Some of these patients develop an immune response to the normal collagen antigens present in the GBM of the donor kidney , and in a small minority anti-GBM disease develops and destroys the allograft.
Immunohistochemistry (GBM) in X-linked Alport’s syndromeNormalmale XLfemale AS
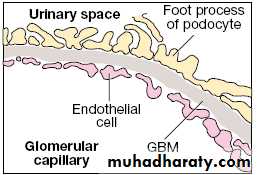
Diagrammatic structure of the normal GBM...
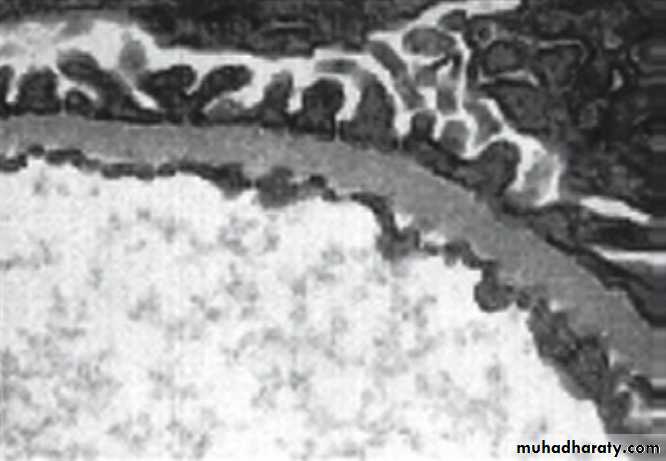
The normal GBM (electron micrograph) contains mostly the tissues pecificα3, α4 and α5 chains of type IV collagen
In Alport’s syndrome this network is disrupted and replaced by α1 and α2 chains. Although the GBM
appears structurally normal in early life, in time thinning appears, progressing to thickening, splitting and degeneration
Ocular abnormalities in Alport’s syndrome. A, Anterior lenticonus shown by slit-lamp ophthalmoscopy. B, Perimacular flecks
Thin GBM disease
There is glomerular bleeding, usually only at the microscopic or dipstick level,Without hypertension, proteinuria or reduction of GFR.
light microscopy The glomeruli appear normal
electron microscopy the GBM is abnormally thin.
This autosomal dominant condition accounts for a large
proportion of ‘benign familial haematuria’
excellent prognosis.
Some families may be carriers of autosomal recessive Alport’s syndrome but this does not account for all cases.
Cystic kidney diseases
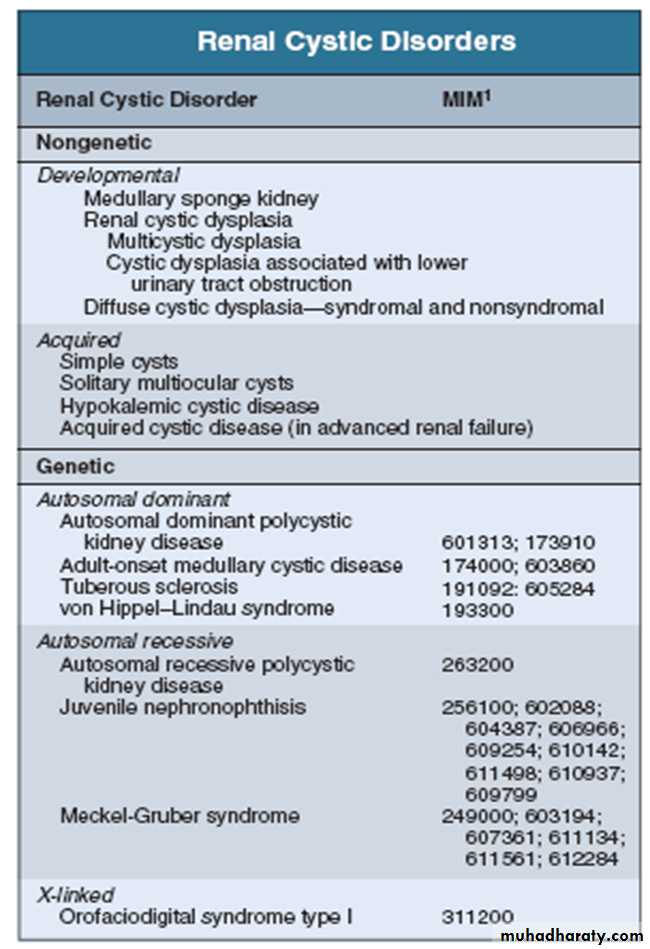
Cystic kidney diseases
Solitary or multiple simple renal cysts ;-are common, with advancing age:-
50 % of > 50 years have one or more such cysts.
No special significance except in the Diff. Dx of renal tumours.
often asymptomatic, accidental finding on U/S exam.
Occasionally they may cause ;-- pain and/or haematuria owing to their large size,
- bleeding may occur into the cyst.
-
Adult polycystic kidney disease AKPD
- APKD a prevalence rate of 1:1000.,.I 3–10 % of all patients on regular dialysis in the West .
Pathology;-
- Small cysts lined by tubular epithelium
- develop from infancy or childhood
- enlarge slowly and irregularly.
- surrounding normal kidney tissue is progressively
attenuated.
- Renal failure is associated with grossly enlarged kidneys .
Adult polycystic kidney disease AKPD
- Inheritance
Autosomal dominant. Mutations in ;-
85% PKD1 gene /chrom 16 , ESRD 50% , onset 50 yrs
15% PKD2 gene / chrom 4 , ESRD minority , 70 yrs
These genetic abnormalities are distinct from the autosomal recessive form of polycystic disease (due to mutations in the PKHD1 gene on chromosome 6 , which is often lethal in early life
Adult polycystic kidney disease AKPD
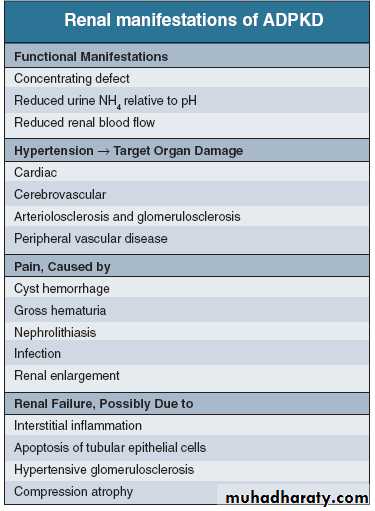
Clinical features- usually asymptomatic until later life. After the age of 20 .
- insidious onset of hypertension. RAS (ACEI of choice)
Early control of BP is essential as cardiovascular
complications are a major cause of death in ADPKD
- Abdominal pain ;-
acute loin pain and/or haematuria owing to haemorrhage into a
cyst, cyst infection or urinary tract stone formation
loin or abdominal discomfort / increasing size of the kidneys
- One or both kidneys may be palpable +/- nodular surface.
- Haematuria (with little or no proteinuria)
- Urinary tract or cyst infections
Adult polycystic kidney disease AKPD
Progressive renal failure
the most serious complication
GFR below 50 mL/min, decline in averages 5 mL/min/year, which is more rapid than in other primary renal disorders
Survival rates on haemodialysis and after renal transplantation in ADPKD are better than other primary renal diseases
Symptoms of uraemia and /or anaemia associated with CRF
PKD is not a pre-malignant conditionAdult polycystic kidney disease AKPD
Associations;-30 % Hepatic cysts ,
- 20% Mitral and aortic valves regurgitation
- 10% Berry aneurysms of brain +/- SAH
- Colonic diverticulae
- Abdominal wall hernias may occur.
MRI images of kidneys. A Normal kidneys. B Polycystic kidneys; although the kidney enlargement is extreme, this patient had only slightlyreduced GFR.
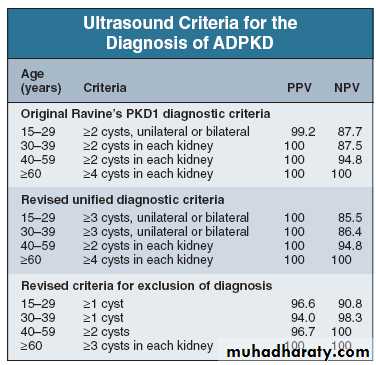
AKPD Investigations and screening
Family Hx, clinical findings & U/S exam.U/S cysts in 95% of patients over 20 years ,
( bilateral, multiple cysts, not just two or three).
not detect small developing cysts in youngers
sometime possible to make a specific genetic diagnosis,
- Screening for intracranial aneurysms is not generally indicated, where non-invasive MR angiography is available, some centers screen patients in families with a history of subarachnoid haemorrhage .
Ultrasound criteria for diagnosis of ADPKD. NPV,negative predictive value; PPV, positive predictive value
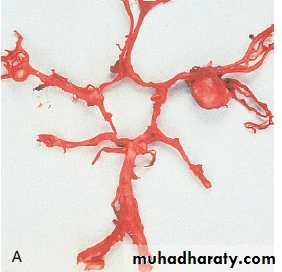
Markedly enlarged polycystic kidneys in comparison to a normal kidney in the middle.
Vascular manifestations of ADPKD. , Gross specimendemonstrating bilateral aneurysms of the middle cerebral arteries
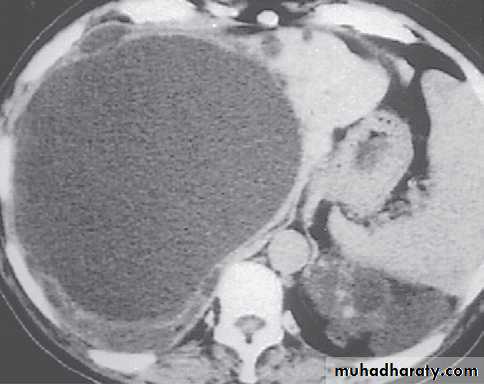
polycystic liver disease. A, a very large, cyst. B several large cysts. C, multiple smaller cysts
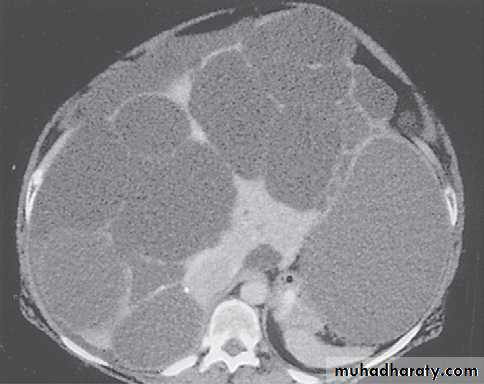
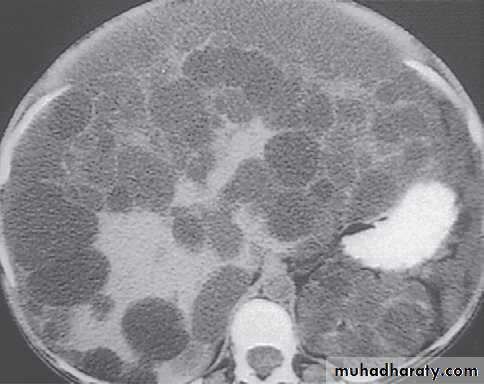
AKPD Management
drugs that seem to slow cyst growthThese therapies include;-
- the vasopressin V2 receptor inhibitor, roscovitine
- anti proliferative therapy with sirolimus
- Good control of BP is important
- Good candidates for dialysis and transplantation.
- Nephrectomy
- large , to make space for a renal transplant.
- source of pain or infection,
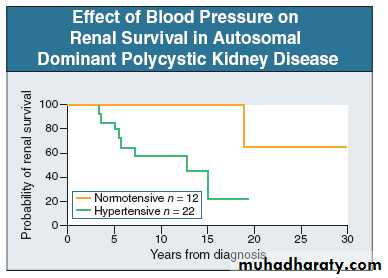
Patients with PKD and HrT at diagnosis have less probability of renal survival than those with normal blood pressure
Other cystic diseases
Medullary sponge kidney- Cysts confined to papillary collecting ducts.
- Cause is unknown ,not inherited
- Patients usually present as adults with renal
stones ,often recurrent, complications
- Prognosis is generally good.
The diagnosis is made by ultrasound or IVU.
Contrast medium is seen to fill dilated or cystic tubules, which are sometimes calcified.
MSK. A, plain film shows medullary nephrolithiases. B, IVU 10-min, clusters of rounded densities in the papillae discrete linear opacities (paintbrush appearance).
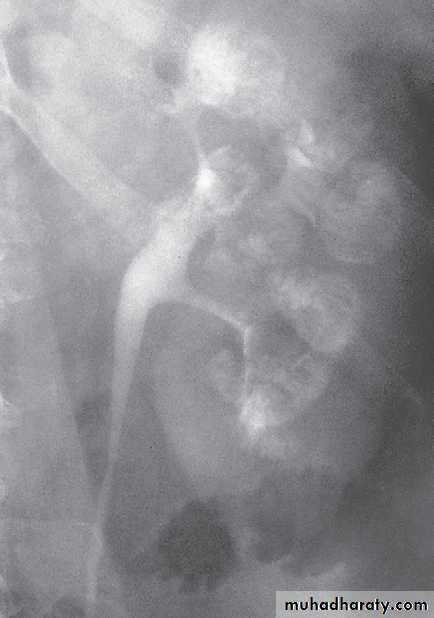
MSK. IVUcontrast medium filling both the collecting system and cavities arising from collecting ducts, The cavities have been likened to bunches of grapes..
MSK Non enhanced CT reveals densely echogenic foci in the medulla.
Medullary cystic kidney diseases
are a heterogeneous group of inherited disorders, known as nephronophthisis in children.
Small cortical cysts are associated with progressive destruction of the nephron.
The childhood variants are characterized by thirst and polyuria due to nephrogenic diabetes insipidus,
often with a family history of similar disease.
Sometimes, affected patients are ‘salt-losing’,
Even when they are treated appropriately, serious renal failure is usual.
?? genetic basis
Other cystic diseases
Acquired cystic kidney diseaseVery long history of renal failure, usually including many years of dialysis or transplanted.
Cystic degeneration (formation of multiple cysts which
enlarge with time)
- associated with increased erythropoietin production
- sometimes they develop malignant tumour formation( renal cell carcinoma) More common than in the general population
Reflux nephropathy (chronic pyelonephritis)
This is a chronic interstitial nephritis associated(VUR) in early life,- with the appearance of ‘scars’ in the kidney, by imaging .
- The incidence about 12%
Pathogenesis
-Susceptibility with genetic component,
- urine refluxes back from the bladder into the ureter,
recurrent UTI in childhood,
- Antenatal U/S ;- renal scars in utero in the absence of infection.
Reflux nephropathy (chronic pyelonephritis)
Pathology
Reflux diminishes as the child grows and usually disappears.
It is often not demonstrable in an adult with a scarred kidney
.
unilateral or bilateral ,.
Gross scarring of the kidneys, commonly at the poles, with reduced size and narrowing of the cortex and medulla.
In patients who develop heavy proteinuria and hypertension, renal biopsies show glomerulomegaly and focal glomerulosclerosis, probably as a secondary response to reduced nephron number and functional mass.
Reflux nephropathy Clinical features
Usually asymptomatic, at any age with hypertension , proteinuria orfeatures of CKD.
- +/- recurrent UTI. and aching lumbar pain., may renal calculi
- Urinary white cells and proteinuria (usually < 1 g/24 hrs)
- first present with hypertension and/or proteinuria in pregnancy.
In some families there is a clear inheritance pattern
Investigations
U/S is an insensitive For detecting renal scars, BUT it will detect major dysplasia , dysgenesis, and exclude significant obstruction.
Radionuclide DMSA scans are more sensitive
imaging by MRI or CT maybe useful.
micturating cysto-urethrography (MCUG)used less often.
Reflux nephropathy &Management
Management
- Infection, treated & prevented with prophylactic ABCS.
reduce recurrences of UTI but there is no evidence that they
protect against further renal scarring or dysfunction.’
nephrectomy - pyelonephrosis
- unilateral renal infection or pain persists,.
- hypertension is cured by when the disease
is predominantly or entirely unilateral.
As most childhood reflux tends to disappea spontaneously
and trials have shown small or no benefits from anti-reflux surgery, such intervention is now less common
Reflux nephropathy &Management
Prognosis- Children and adults with small or unilateral renal scars have a good prognosis, provided renal growth is normal.
- With significant unilateral scars / usually compensatory hypertrophy of the contralateral kidney.
In patients with more severe bilateral disease, prognosis is predicted by the severity of renal dysfunction , hypertension and proteinuria.
If the serum creatinine is normal and hypertension and proteinuria are absent, then the long-term prognosis is usually good.
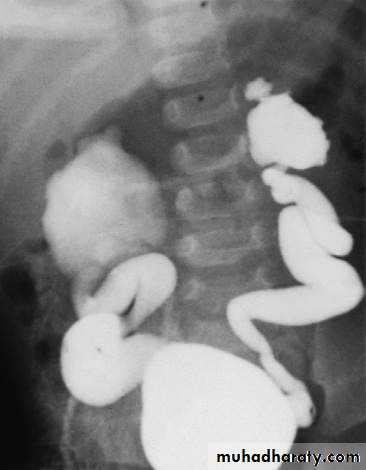
VUR (grade IV) shown by micturating cystogram. The bladder has been filled with contrast medium through a urinary catheter. After micturition there was gross VUR into widely distended ureters and pelvicalyceal systems.