Disorders of Hemostasis 5th Year Medical Students
Dr. salma al-hadadApril 18th 2016
Objectives
To describe the bleeding manifestation; various clinical presentation, laboratory tests and diagnosis.To list the causes of thrombocytopenia
To outline the steps of diagnosis of Idiopathic Thrombocytopenic Purpura (ITP)
To describe the clinical manifestation and diagnosis of Hemophilia, Von Willebrand Disease & Henoch- Schonlein Purpura
Normal Hemostasis
It is the active process that clots blood in areas of blood vessel injury, yet simultaneously limits the clot size only to the areas of injury.As a result of injury to the blood vessel endothelium, three events take place simultaneously:
• Vasoconstriction - (vascular phase)
• Platelet plug formation (primary haemostatic mechanism— (platelet phase)
• Fibrin thrombus formation (secondary haemostatic mechanism — (plasma phase)
Hemostatic Failure
Inappropriate and excessive bleeding either spontaneous or in response to injury
Bleeding Manifestations
In disorders of hemostasis the bleeding manifestations are commonly present at more than one site.• Spontaneous skin bruising or purpura
• Bleeding from mucous membranes, e.g. nose/mouth, GIT, urinary & genital tract
• Bleeding from venepuncture sites, IV cannulation, operation sites and from tooth sockets post dental extraction
• Bleeding into muscles, joints or deep tissues
• Menorrhagia
• Cerebral hemorrhage
Clinical Evaluation of Bleeding Patients
“80% of correct diagnosis can be made by history taking and physical examination”
History Taking
Identify if the bleeding problem is due toLocal vs. systemic defect
Location: single vs. multiple sites
Severity: Spontaneous? Appropriate to trauma?
Hereditary vs. acquired disorder
Onset
Family history
Underlying disease
Medication
Primary vs. secondary hemostatic disorder
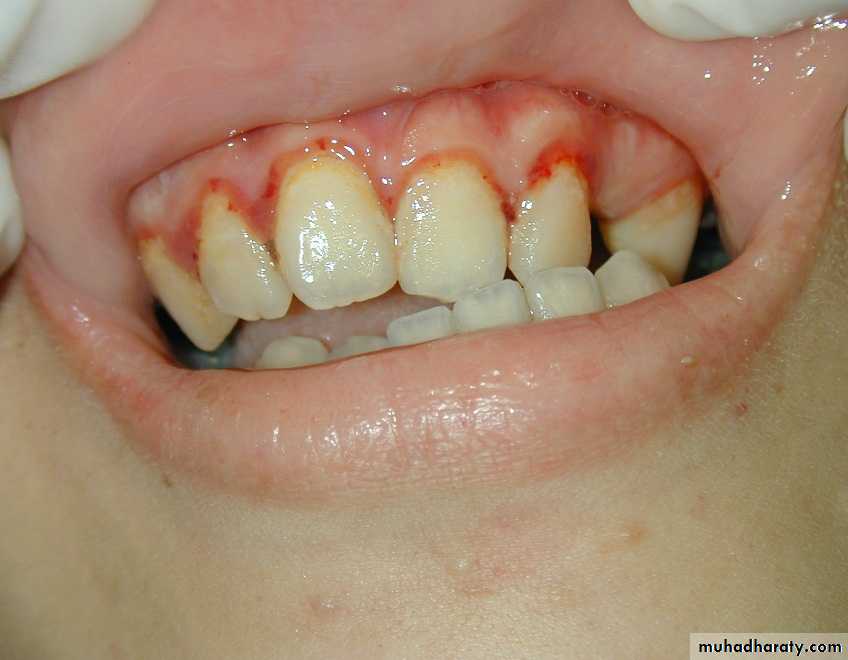
Primary Hemostatic defect
Secondary Hemostatic defect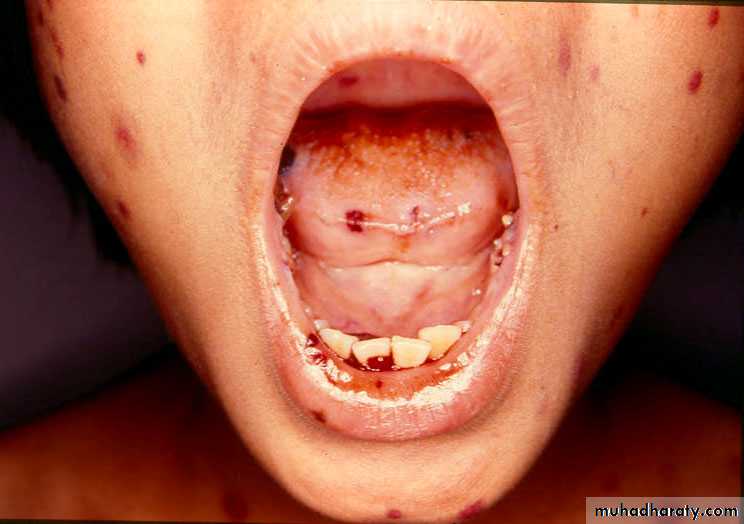
Clinical manifestation of the bleeding
• Mucosal bleeding; easy bruises, epistaxis, menorrhagia, petechae, and oozing from surgical wounds is most consistent with a defect in primary hemostasis. Mostly due to defects in platelets, Von Willebrand Factor, or the vessel wall.• Deep tissue bleeding; (hematomas, joint and muscle hemorrhages) and “delayed” surgical bleeding are more suggestive of a coagulation factor abnormality, e.g. hemophilia.
Petechae
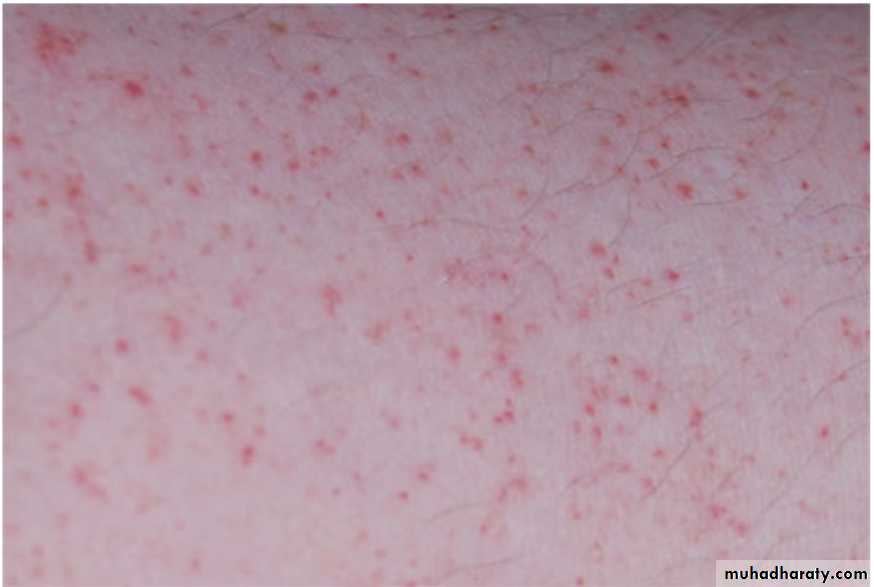

Do not blanch with pressure, Not palpable
(typical of platelet disorders)
Clinical Manifestations of Hemostasis
• Clinical characteristic• Platelets defect
• Clotting factor deficiency
• Site of bleeding
• Skin, mucous membranes (gingival, nares, GI and genitourinary tracts)
• Deep in soft tissues (joints, muscles)
• Bleeding after minor cuts
• Yes
• Unusual
• Petechae
• Present
• Absent
• Ecchymoses
• Small, superficial
• Large, palpable
• Hemarthroses, muscle hematomas
• Rare
• Common
• Bleeding after surgery
• Immediate, mild
• Delayed, severe
Clinical Evaluation of Hemostasis
The history should determine:
Site or sites of bleeding
Duration of hemorrhage
Age at onset
Severity was the bleeding spontaneous, or did it occur after trauma, did the symptoms correlate with the degree of injury or trauma?
Was there a previous personal or family history of similar problems?
Clinical Evaluation of Hemostasis
If a child has had surgery affecting the mucosal surfaces, e.g. tonsillectomy or major dental extractions, the absence of bleeding usually rules out a hereditary bleeding disorderHistory of Circumcision in males
It is important to take a careful menstrual history
Drugs: e.g. Anticoagulants, NSAID & Cytotoxics
Laboratory Tests
A reliable laboratory approach, including:First-line (screening) and
second-line (specific) testing,
• Essential to screen, diagnose & monitor patients
First-line (screening) tests:
• CBC: with peripheral smear• Bleeding time: bleeding usually stops within 4–8 min, this test should be done only when platelets count is normal to exclude functional defect
• Platelet function analyzer(PFA-100): to evaluate the platelet functions and VWF interaction
• PT is a measure of the extrinsic (FVII) and common pathway (FV, FX, prothrombin, fibrinogen) clotting factors.
• PTT measures the contact system (prekallikrein, FXII) as well as the intrinsic (FVIII, FIX, FXI) and common pathway clotting factors
• TT measures fibrinogen deficiency
Second-line (specific) tests:
Clotting factor assays
Bleeding time test
IVY method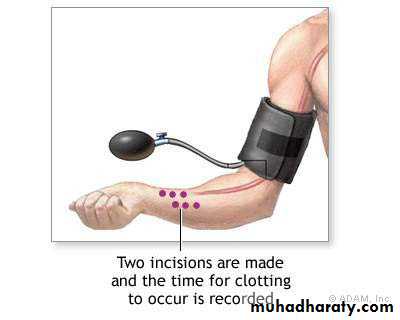
Duke’s test

Laboratory evaluation of bleeding disorders
CBC-complete blood count, F-factor, PFA-platelet function analyzer, PT-prothrombin time, PTT-activated partial thromboplastin time, RIPA-ristocetin-induced platelet aggregation, vWD-von Willebrand disease, vWF-von Willebrand factor, vWF:Rco-ristocetin cofactor activity.Platelet abnormalities- Quantitative
• Decreased bone marrow production:• a- Malignant marrow infiltration
• b- Drugs
• c- Severe megaloblastic anemia
• d- Hypoplastic anemia
2. Decreased platelet survival (peripheral consumption):
• a- Immune mechanisms:
• Primary —immune thrombocytopenia (ITP)
• Secondary—SLE, lymphoma
• Drugs—Thiazides, Sulfonamides
• b- Excessive consumption:
• Disseminated intravascular coagulation (DIC), splenomegaly (hypersplenism)
Platelet abnormalities- Qualitative
• Inherited: e.g. Bernard Soulier syndrome, thrombasthenia
• Acquired: e.g. myeloproliferative diseases, uremia, Drugs, e.g. aspirin and NSAID.
Case 1
4 yr old boyURTI 2 weeks ago
Sudden onset bruising/petechiae
Past Hx.: Nil
Family Hx.: Nil
Physical examination:
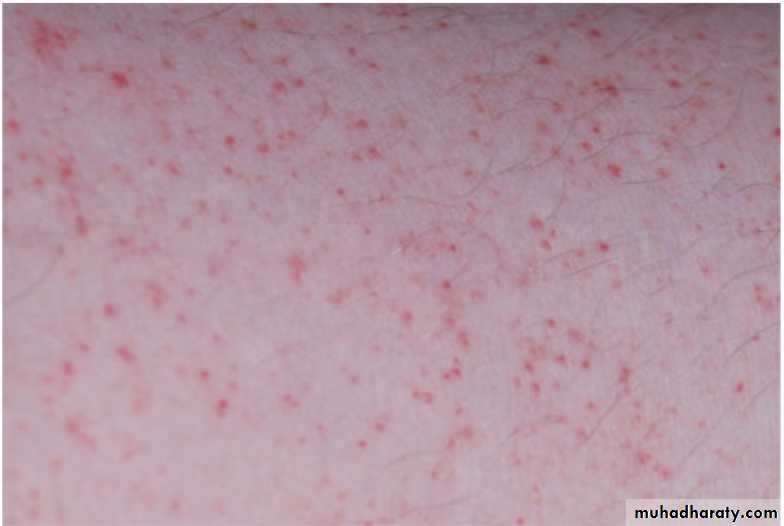
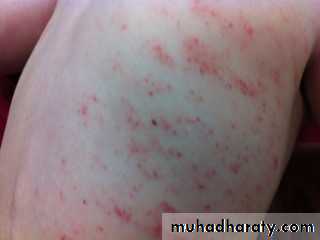
Investigations
Hb 11g/dl; WBC 8.000/cmm; Plat. 35.000/cmm.
PT 14 sec ; PTT 33 sec; Fibrinogen 2.0g/l
Treatment options: Nil
Outcome: 80% recovery; 20% chronic
Case 2
You are on call for ENT and are asked to see a 14-year old girl with sudden refractory nosebleed.The nose is packed & bleeding does not stop.
You noticed a few bruises
The lab results showed with a “critical” platelet count of 10.000/cmm
• What is likely diagnosis?
• What to do?
• What is needed for diagnosis?
• Does she require hospitalization ?
ITP (Immune Thromboctopenic Purpura)
• When isolated and very low--- ITP is most likely diagnosis• If mucosal bleeding &/or platelets are less than 20.000-30.000/cmm, needs action:
Hospitalization
Steroids
IVIG
Anti D
• Bone marrow examination
(ITP) Idiopathic Thrombocytopenic Purpura
Bleeding disorder characterized by isolated low platelet count (Plts. < 130 - 150 x 109/L)
The most common cause of acute onset of thrombocytopenia in an otherwise well child.
1–4 wk after exposure to a common viral infection, an autoantibody directed against the platelet surface develops
Antiplatelet antibody that binds to the platelet surface and enhances its destruction in the spleen and liver
Clinical Manifestations
The classic presentation is that of a previously healthy child who has sudden onset of generalized petechae and purpuraOften there is bleeding from the gums and mucous membranes, particularly with profound thrombocytopenia (platelet count <10 × 109/L).
Physical exam is normal, other than the finding of petechae and purpura.
Absence of HSM or remarkable LAP (which might suggest other diagnoses like leukemia)
Clinical Manifestations
No symptomsMild: bruising and petechae, occasional minor epistaxis, very little interference with daily living
Moderate: more severe skin and mucosal lesions, more troublesome epistaxis and menorrhagia
Severe: bleeding episodes—menorrhagia, epistaxis, melena—requiring transfusion or hospitalization, symptoms interfering seriously with the quality of life
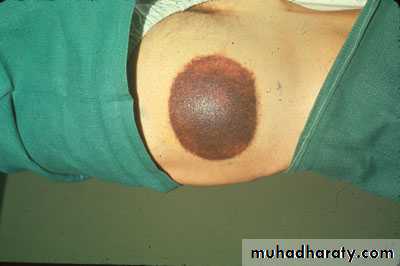
Petechae and Purpura
Petechae & Ecchymoses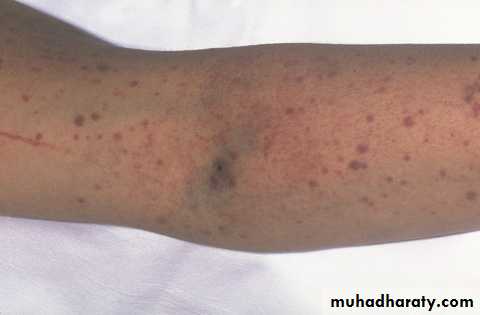
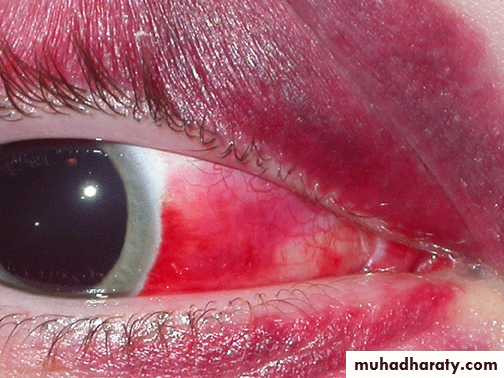
Subconjunctival Hemorrhage
Relationship Between Platelet Count & Bleeding
• Platelet Count• (109/L)
• Signs and Symptoms
• >100
None
50 to 100
Minimal (after major trauma & surgery)
• 20 to 50
Mild (cutaneous)
5 to 20
Moderate (cutaneous and mucosal)
<5
Severe (mucosal and CNS)
Laboratory Findings
Common finding is platelet count <20 × 109/LHb, WBC and differential count should be normal
BM examination is normal (normal or increased megakaryocytes).
Indications for BMA include:
An abnormal WBC or differential
Unexplained anemia
Findings suggestive of bone marrow disease on history & physical examination.
ITP is a diagnosis of exclusion
History: careful drug history
Examination: healthy appearing child, no HSM, no LAP, has petechae, purpura and occasionally mucous membrane bleeding.
Blood counts: CBC should be normal except thrombocytopenia
Peripheral smear evaluation: essential to
• Rule out platelets clumping
• Evaluate WBC and RBC morphology
• Evaluate size of platelets
General Considerations for Initial Management
The goal of all treatment is to achieve an adequate hemostasis (>20 ×109/L) & prevent the rare intracranial hemorrhage, rather than a normal platelet count.The majority of patients with no bleeding or mild/moderate bleeding can be treated with observation alone regardless of platelet count.
First-line treatment includes:
Observation,
Corticosteroids,
IVIG, or
anti-D immunoglobulin.
Platelets transfusions should be reserved to life threatening bleeding and CNS hemorrhage only as their half life is very short in patient with ITP
Prognosis of acute ITP
In 70–80% of children who present with acute ITP, spontaneous resolution occurs within 6 months<1% of patients have intracranial hemorrhage.
20% of children go on to have chronic ITP.
Coagulation factor deficiency
Congenital
Usually single factor deficiencies.
Sometimes clinically apparent at birth, but mild deficiencies may not become apparent until adolescence or adult life,
e.g. Hemophilia A (Factor VIII) and B (Factor IX, Christmas disease), Von Willebrand disease, Factor XI deficiency
Coagulation factor deficiency
• Acquired• Decreased production: e.g. liver disease, Vitamin K deficiency –neonates, malabsorption
• Increased consumption: DIC
• Circulating inhibitors: e.g. antibodies –especially to F. VIII and in SLE
• Drugs: Heparin and Warfarin.
• Dilution: massive, rapid blood transfusion
FVIII or FIX Deficiency (Hemophilia A or B)
Deficiencies of factors VIII and IX are the most common severe inherited bleeding disorders.Clinical Manifestations
Neither factor VIII nor factor IX crosses the placenta; bleeding symptoms may be present from birth.About 2% of neonates with hemophilia sustain intracranial hemorrhages and 30% of male infants with hemophilia bleed with circumcision.
Obvious symptoms of easy bruising, intramuscular hematomas, and hemarthrosis begin when the child “begins to cruise.”
HEMOPHILIA
Non-carrier Mother + Father with Hemophilia
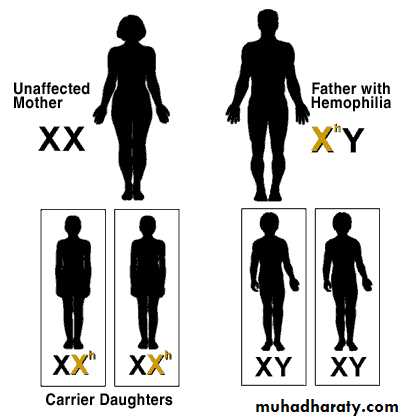
HEMOPHILIA
Carrier Mother + Non-hemophiliac Father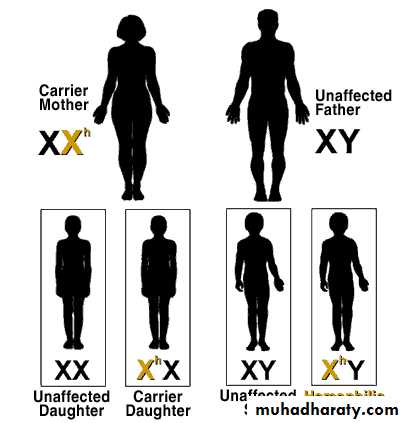
HEMOPHILIA
Carrier Mother + Father with Hemophilia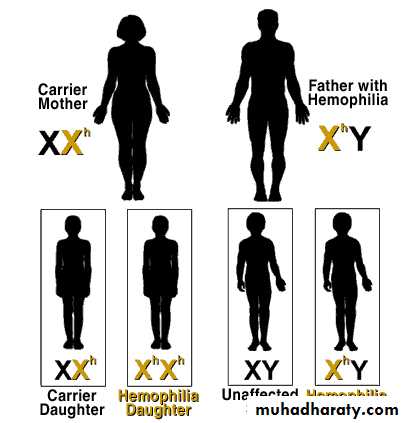
HEMOPHILIA
Mother with Hemophilia + Father with Hemophilia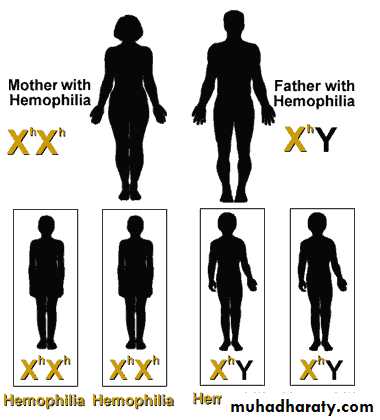
HEMOPHILIA
Mother with Hemophilia + Non-hemophiliac Father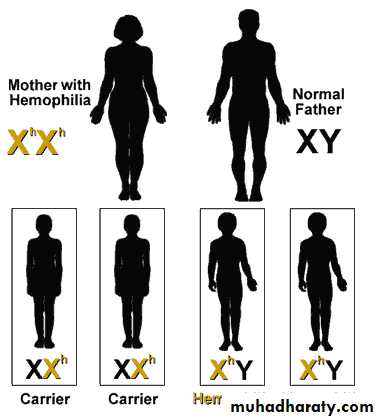
Clinical Manifestations
The hallmark of hemophilia is hemarthrosisBleeding into the joints may be induced by minor trauma; many are spontaneous.
Life-threatening bleeding in the patient with hemophilia is caused by bleeding into vital structures (CNS, upper airway, GIT, or ilio-psoas hemorrhage)
Laboratory Findings & Diagnosis
PTT prolonged in FVIII or FIX deficiencyPlatelet count, bleeding time, prothrombin time, and thrombin time are normal
Specific assay for factors VIII and IX will confirm the diagnosis of hemophilia
Treatment
Early, appropriate therapy is the hallmark of excellent hemophilia care.• Factor VIII or IX concentrate:
When mild to moderate bleeding occurs, levels of FVIII or FIX must be raised to hemostatic levels in the 35–50% range
For life-threatening or major hemorrhages, the dose should aim to achieve levels of 100% activity
Treatment
• Intranasal Desmopressin in mild hemophilia A, it is not effective in hemophilia B• Prophylaxis treatment: recombinant FVIII or IX products, it was recently started aiming to be the standard of care for most children with severe hemophilia to prevent spontaneous bleeding and early joint deformities
Supportive Care
Advise parents that their child should avoid trauma
Avoid violent contact sports
Early psychosocial intervention helps the family to achieve a balance between overprotection and permissiveness.
Avoid Aspirin and NSAIDs that affect platelet function.
Receive the vaccinations against hepatitis B, even with the use of recombinant products
Patients exposed to plasma-derived products should be screened periodically for hepatitis B and C, HIV, and LFT.
Chronic Complications
Long-term complications of hemophilia A and B include:• Chronic arthropathy
• The development of an inhibitor to FVIII or FIX.
• The risk of transfusion-transmitted infectious diseases.
Education remains crucial in hemophilia care
Arthropathy
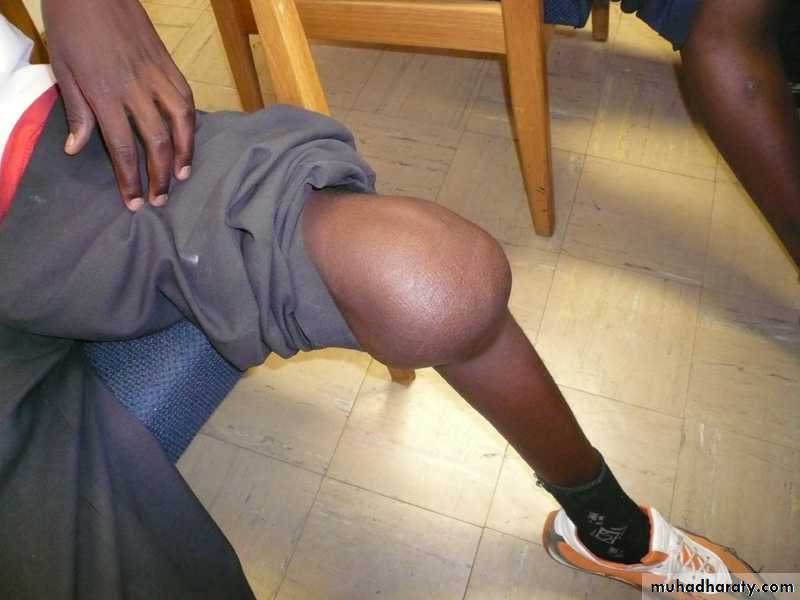
Case 3
14 year old girl with menorrhagiaHistory of easy bruising
CBC normal
PTT 32 (2 sec prolonged)
What is diagnosis?
How to diagnose?
Treatment?
Von Willebrand’s Disease
Most frequent inherited bleeding disorder affect 1-2% of general population
less severe than hemophilia
Disease results from a decrease or absence of Von Willebrand factor required for platelet adhesion
Affects primary hemostasis
Clinical features of VWD
Generally mild bleeding - often unrecognized until surgery or injuryepistaxis, menorrhagia, easy bruising, dental and post operative bleeding
Can be severe in certain types
Requires accurate diagnosis
Requires specific treatment
VWD -types
Type Imost frequent, quantitative defect (decreased VWf )
Type II
qualitative defect (abnormal VWf )
Type III
severe, rare, (absence of VWf )
Laboratory Findings
Long BT and a long PTT
Normal results on screening tests do not exclude the diagnosis of VWD
if the history is suggestive of a muco-cutaneous bleeding disorder, VWD testing should be undertaken
Treatment
It is directed toward increasing the plasma level of VWF & FVIII.Current replacement therapy uses plasma-derived VWF containing concentrates that also contain factor VIII.
Purified or recombinant VWF concentrates (containing no factor VIII) may become available in the near future
Dental extractions and sometimes nosebleeds can be managed with both DDAVP & anti fibrinolytic agent
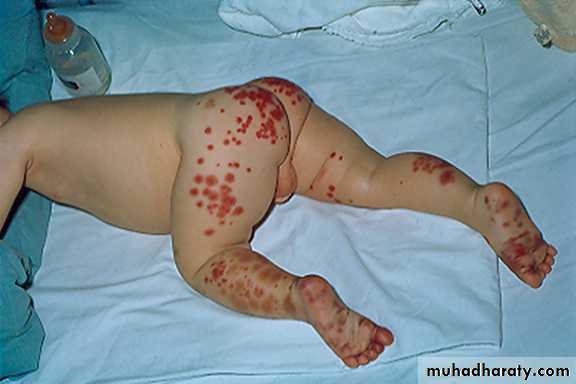
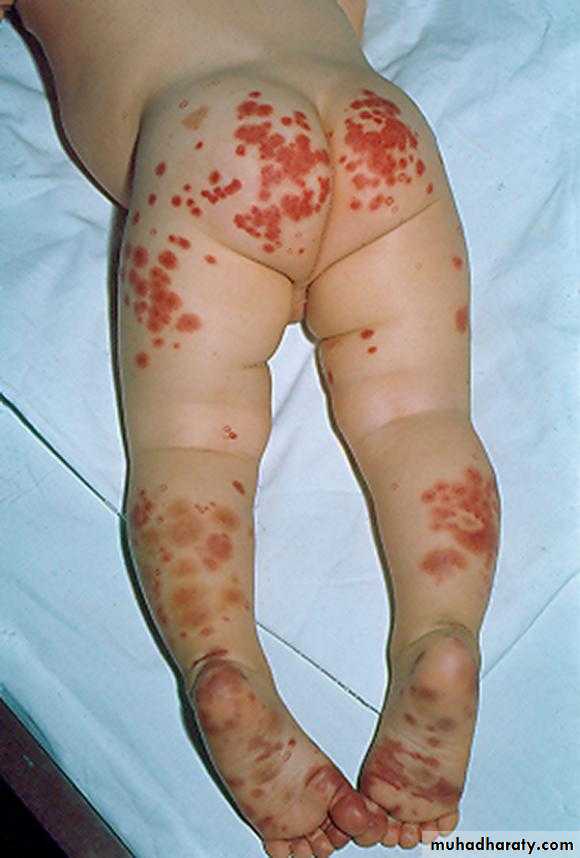
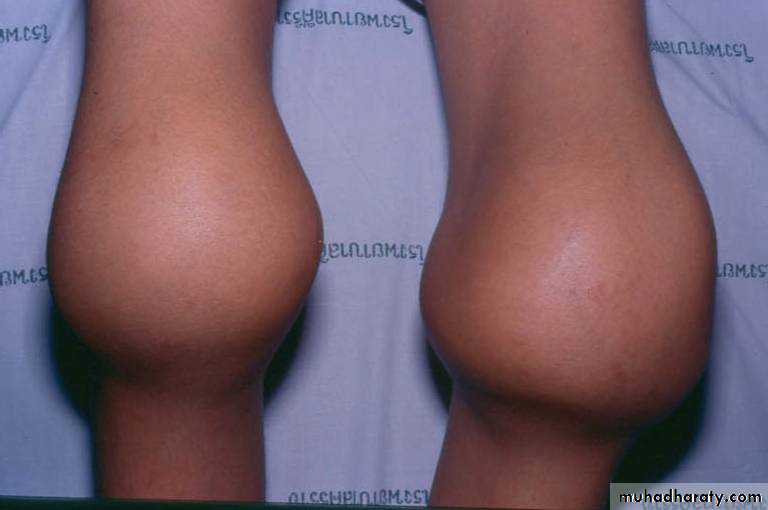
Henoch-Schönlein Purpura
Sudden development of a purpuric rash, arthritis, abdominal pain, and renal involvement.The characteristic rash, consisting of petechae & often palpable purpura, usually lower extremities & buttocks.
Coagulation studies are normal
The pathologic lesions in the skin, intestines, and synovium, inflammatory damage to the endothelium of the capillary mediated by WBC & macrophages (Vasculitis)
The trigger for HSP is unknown. In the kidney, there is focal GN with deposition of immunoglobulin A.
Henoch-Schönlein Purpura